Phase I/II Study of Safety and Preliminary Efficacy of Intravenous Allogeneic Mesenchymal Stem Cells in Chronic Stroke

Authors:
Michael L. Levy, MD, PhD; John R. Crawford, MD; Nabil Dib, MD; Lev Verkh, PhD; Nikolai Tankovich, MD, PhD; Steven C. Cramer, MD
Background and Purpose
Stroke is a leading cause of long-term disability. Limited treatment options exist for patients with chronic stroke and substantial functional deficits. This study examined the safety and preliminary efficacy of intravenous allogeneic mesenchymal stem cells in this population.
Methods
Entry Criteria
- Ischemic stroke >6 months prior
- Substantial impairment (National Institutes of Health Stroke Scale score ≥6)
- Disability
Treatment Procedure
- Enrollees received a single intravenous dose of allogeneic ischemia-tolerant mesenchymal stem cells.
- Phase 1 used a dose-escalation design (3 tiers, n=5 each).
- Phase 2 was an expanded safety cohort.
Primary and Secondary End Points
- The primary end point was safety over 1 year.
- Secondary end points examined behavioral change.
Results
Phase 1 Findings (n=15)
- Each dose (0.5, 1.0, and 1.5 million cells/kg body weight) was found safe.
Phase 2 Findings (n=21)
- Subjects received 1.5 million cells/kg.
- At baseline, subjects (n=36) averaged 4.2±4.6 years post-stroke, age 61.1±10.8 years, National Institutes of Health Stroke Scale score 8 (6.5–10), and Barthel Index 65±29.
- Two were lost to follow-up, one was withdrawn, and two died (unrelated to study treatment).
Adverse Events
- Of 15 serious adverse events, none was possibly or probably related to study treatment.
- Two mild adverse events were possibly related to study treatment: a urinary tract infection and intravenous site irritation.
- Treatment was safe based on serial exams, electrocardiograms, laboratory tests, and computed tomography scans of chest/abdomen/pelvis.
Behavioral End Points
- All behavioral end points showed significant gains over the 12 months of follow-up.
- Barthel Index scores increased by 6.8±11.4 points at 6 months (P=0.002) and by 10.8±15.5 points at 12 months (P<0.001) post-infusion.
- The proportion of patients achieving excellent functional outcomes (Barthel score ≥95) increased from 11.4% at baseline to 27.3% at 6 months and to 35.5% at 12 months.
Conclusions
Intravenous transfusion of allogeneic ischemia-tolerant mesenchymal stem cells in patients with chronic stroke and substantial functional deficits was safe and suggested behavioral gains. These data support proceeding to a randomized, placebo-controlled study of this therapy in this population.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov
Unique identifier: NCT01297413
Key Words: abdomen, brain ischemia, neuroprotection, pelvis, reperfusion
Mesenchymal Stem Cells as Restorative Therapy for Stroke Patients
Stroke is perennially among the leading causes of human disability and the leading neurological cause of lost disability-adjusted life years. The mean survival after stroke is 6 to 7 years, and indeed more than 85% of patients live past the first year poststroke, many with years of enduring disability. Many restorative therapies are under study to improve outcomes after stroke. Restorative therapies aim to improve patient outcomes by promoting the neural processes underlying behavioral recovery and are distinguished from acute therapies, such as reperfusion or neuroprotection, that aim to reduce initial injury. As such, restorative therapies often have a time window measured in days to months, or in some cases, years.
Mesenchymal Stem Cells (MSC) as a Candidate for Stroke Therapy
Mesenchymal stem cells (MSC), also known as mesenchymal stromal cells, are among the leading restorative therapy candidates. Substantial preclinical data support the safety and efficacy of MSC as a restorative therapy to improve outcomes after stroke. For example, a meta-analysis reported that 44 of 46 preclinical stroke studies found MSC to be superior to placebo, with effect sizes greater than 1.0.
Initial Human Studies of MSC
Initial human studies of MSC (or MSC-like cells) after stroke focused on autologous cell therapies, whereby bone marrow is taken from each patient to produce his/her own MSC batch, and found MSC infusion to be safe. MSC are relatively immunoprivileged given their very low levels of human leukocyte antigen molecule expression, a fact that opens the door to the administration of allogeneic MSC. Allogeneic MSC have been found to be safe without the use of concomitant immunosuppression, and can be manufactured in a manner that enables broad clinical application.
Studies of Allogeneic MSC
Studies of allogeneic MSC (or MSC-like cells) poststroke have focused on early time points (administration 24–48 hours poststroke) or used an invasive procedure to implant cells intracerebrally. Each approach has its relative advantages and disadvantages, and an intravenous method of introducing MSC, if comparably efficacious, might facilitate widespread implementation and also avoid adverse events attributable to invasive procedures.
Current Study Overview
The current study was a phase I/II dose-escalation trial that examined the effects of a single intravenous infusion of allogeneic ischemia-tolerant MSC. The target population was patients with chronic ischemic stroke and substantial functional deficits, a group for whom treatment options remain limited. The primary outcome was safety, based on serial measures of behavior, computed tomography (CT) scans, and laboratory testing. Preliminary estimates of treatment efficacy were also examined.
Inclusion Criteria
- Age ≥18 years
- Ischemic stroke ≥6 months prior, radiologically confirmed at initial diagnosis and at study enrollment
- Severe disability resulting from the index stroke, operationally defined as subject confined to a wheelchair, requiring home nursing care, or needing assistance with activities of daily living
- No substantial improvement in neurological or functional status for the 2 months before study enrollment
- NIHSS score 6–20
- Life expectancy >12 months
- Patient receiving standard of care secondary stroke prevention before enrollment
- Patient or a surrogate able to provide informed consent
- Reasonable expectation that the patient will receive standard posttreatment care and attend all scheduled study visits
- Adequate systemic organ function, specifically:
- Serum aspartate aminotransferase ≤2.5× upper limit of normal
- Serum alanine aminotransferase ≤2.5× upper limit of normal
- Total serum bilirubin ≤1.5× upper limit of normal
- Prothrombin time and partial thromboplastin time ≤1.25× upper limit of normal in subjects not receiving antithrombotic therapy
- Serum albumin ≥3.0 g/dL
- Absolute neutrophil count ≥1500/µL
- Platelet count ≥150,000/µL
- Hemoglobin ≥9.0 g/dL
- Serum creatinine ≤1.5× upper limit of normal
- Serum amylase or lipase ≤1.0× upper limit of normal
Exclusion Criteria
- History of uncontrolled seizure disorder
- History of cancer within the past 5 years, except for localized basal or squamous cell carcinoma
- History of cerebral neoplasm
- Positive for hepatitis B, C, or HIV
- Myocardial infarction within 6 months of study entry
- Presence of any other clinically significant medical or psychiatric condition, or laboratory abnormality, for which study participation would pose a safety risk in the judgment of the Investigator or Sponsor
- Findings on baseline computed tomography suggestive of subarachnoid or intracerebral hemorrhage within the past 12 months
- Participation in another investigational drug or device study in the 3 months before treatment
- History within the past year of drug or alcohol abuse
- Pregnant or lactating, or expectation to become pregnant during the study
- Allergy to bovine or porcine products
NIHSS indicates National Institutes of Health Stroke Scale.
Methods
Study Design
This was a phase I/II multi-center, open-label study that aimed to evaluate the safety and preliminary efficacy of a single intravenous infusion of marrow-derived allogeneic ischemia-tolerant MSC. Entry criteria appear in Table 1 and in sum describe enrollment of adults with radiologically verified chronic stable ischemic stroke and substantial impairment and functional deficits. Patients were followed for one year after MSC infusion. The study made no restrictions on, and did not provide any forms of, medication or therapy (occupational, physical, or speech) during the follow-up year after infusion. All patients signed consent in accordance with local Institutional Review Board approval. This study was approved by the Food and Drug Administration and was registered at clinicaltrials.gov. The data that support the findings of this study are available from the corresponding author on reasonable request.
The study occurred in 2 parts, with part 1 being a dose-escalation study and part 2 being an expanded safety study based on part 1 findings. Part 1 consisted of 3 cohorts (n=5 per cohort) enrolled sequentially in a dose-escalation manner, with subjects receiving one of 3 doses based on body weight, with a maximum dosage of 150 million cells. Cohort 1 received 0.5 million cells/kg of body weight; Cohort 2, 1.0 million cells/kg; and Cohort 3, 1.5 million cells/kg. The dose-escalation plan in part 1 required a review by the Data Safety Monitoring Board once the 5 subjects in Cohort 1 were treated and evaluated through study day 10. If safety was established, Cohort 2 was to proceed at the next highest dose, followed by a similar safety review before escalation to the highest dose in Cohort 3. Part 2 aimed to enroll an additional minimum of 20 subjects at the highest safe dose level determined in part 1. An additional interim review was conducted by the Data Safety Monitoring Board after the first 5 patients were treated in part 2. Detailed stopping rules appear in the online-only Data Supplement (see Stopping Rules and Determination of Maximum Tolerated Dose).
The target dose of 1.5 million cells/kg corresponds to allometric scaling from animal studies. Our meta-analysis of preclinical studies of MSC after experimental ischemic stroke identified 9 rodent studies that transfused MSC using the intravenous route in the post-acute period. In each study, MSC provided substantial behavioral gains (effect sizes >1.0), using doses ranging from 3.6 to 12.4×10^6 MSC/kg body weight (mean dose of 10.1×10^6 MSC/kg). The approach to allometric scaling from animals to humans recommended by the Food and Drug Administration uses a body surface area normalization, which for the mean value in rodents yields a comparable human dose of 1.6×10^6 MSC/kg.
Cell Manufacturing and Shipping
Manufacturing of MSC was performed at the GMP-compliant facility of the sponsor, Stemedica Cell Technologies, Inc (San Diego, CA). MSC were grown from the bone marrow of a single human donor and are from the same batch used in prior preclinical and clinical studies. Cells were grown under low oxygen (5%) conditions. Such ischemia-tolerant MSC have advantages compared with those grown under normoxic conditions, for example, showing higher proliferation rate, expression of stem cell-related genes, production of key cytokines, and migration activity. Cells were harvested at passage 4 and expressed CD105, CD73, and CD90 surface markers, consistent with the International Society for Cellular Therapy definition. Cells were cryopreserved by suspending in Cryostar CS10 freezing medium (BioLife Solutions, Bothell, WA) then stored in the vapor phase of liquid nitrogen. This parent cell bank was then tested for quality control including cell count, viability, appearance, and quantitative polymerase chain reaction for viruses including HIV, Epstein-Barr virus, cytomegalovirus, hepatitis B virus, parvovirus B19, and hepatitis C virus. Cryovials were shipped at ≤-150°C in a vapor phase liquid nitrogen shipper with temperature monitor.
Infusion of Investigational Product
Each site’s pharmacy prepared MSC for infusion per a study-provided protocol. Cryovials (the number of which was based on the dose to be infused) were thawed and MSC were washed in, and then suspended in, Lactated Ringer’s solution at a concentration of 1×10^6 cells/mL using one to three 60 mL syringes. The suspension then underwent final testing before being released for intravenous infusion, consisting of cell count, endotoxin, Gram stain, and review of appearance. Cell count was performed using 0.1% Trypan Blue and a hemacytometer, which also yielded % cell viability. The minimum percent cell viability was required to be ≥70% for the cells to be released. A sample was also sent for subsequent sterility testing. After release by the pharmacy, the final formulation was stored at 2° to 8°C and infused within 8 hours of preparation.
MSC Administration
Before MSC infusion, a 0.1 mL aliquot of the final MSC formulation was injected intradermally; any subject showing a positive reaction (e.g., wheal with erythema) would not be infused. Cells were administered intravenously via metered-dose syringe pump at 2 mL/min. Patients remained in the inpatient telemetry unit for observation until clinically stable.
Patient Assessments
Patients had frequent monitoring until discharged from the telemetry unit. After discharge, patients had safety evaluations on day 2, 3, 4, and 10, then again on month 1, 3, 6, 9, and 12. Adverse events were coded according to the MedDRA adverse event dictionary. The relationship that adverse events had to the investigational product was assessed by the site investigator. Patients were followed for one year using tests of behavior, serology, blood chemistry and cell counts, electrocardiogram, urine, and CT of chest, abdomen, and pelvis. The full schedule of assessments appears in Table SI in the online-only Data Supplement.
Statistics
The primary study endpoint was safety and tolerability, evaluated in all subjects who received any portion of an infusion, and determined by the incidence/severity of adverse events, clinically significant changes on laboratory and imaging tests, vital signs, and physical plus neurological examinations. Four secondary endpoints were scored serially to derive preliminary estimates of efficacy: National Institutes of Health Stroke Scale, Barthel Index (BI), Mini-Mental Status Exam, and Geriatric Depression Scale. For each, the change from baseline was evaluated using the Wilcoxon signed-rank test, with primary analysis of preliminary efficacy being change from baseline to 6 months post-infusion, and analysis including all subjects who received an infusion except for one subject who failed to return after the day 10 visit for all visits (except for month 9 follow-up). For any subject missing 6-month data, 9-month or 12-month data were substituted for this analysis, otherwise missing data were not imputed. Data were analyzed using R statistical software. Given the exploratory nature of this study, sample size was selected as appropriate for detection of any safety concerns in an early phase clinical trial.
Results
Subjects
Of 50 subjects who seemed eligible on prescreening, 36 were enrolled and received treatment from March 14, 2011, to December 15, 2016 (Figure and Table 2). There were 13 subjects enrolled at the University of California, San Diego, 19 subjects at Arizona, and 4 subjects at the University of California, Irvine. Interim safety reviews disclosed no concerns, and so 5 subjects received 0.5×10^6 cells/kg in part 1/Cohort 1, 5 subjects received 1.0×10^6 cells/kg in part 1/Cohort 2, 5 subjects received 1.5×10^6 cells/kg in part 1/Cohort 3, and all 21 subjects in part 2 received 1.5×10^6 cells/kg. For the 15 subjects in part 1, 12 completed the study, 2 died of unrelated causes (coronary artery disease 6 months post-infusion and sepsis 1 month after infusion), and 1 was lost to follow-up after day 10 (reappearing only for the month 9 follow-up visit). For the 21 subjects in part 2, 19 completed the study, 1 was lost to follow-up after month 6, and 1 was withdrawn by the site PI after month 6 due to treatment with another investigational product. Of the 36 subjects enrolled, the planned dose was delivered within 2 mL (i.e., within 2×10^6 cells) of the target in 26 subjects, whereas in 10 subjects a median of 7.6 (interquartile range, 4.4–10.25) mL (i.e., 7.6×10^6 cells) was not infused as planned, which represented a median of 6.5% (5.3–9.8) of the intended dose. A total of 179 protocol deviations were reported, mainly related to scheduling study visits or study testing (Table SII in the online-only Data Supplement).
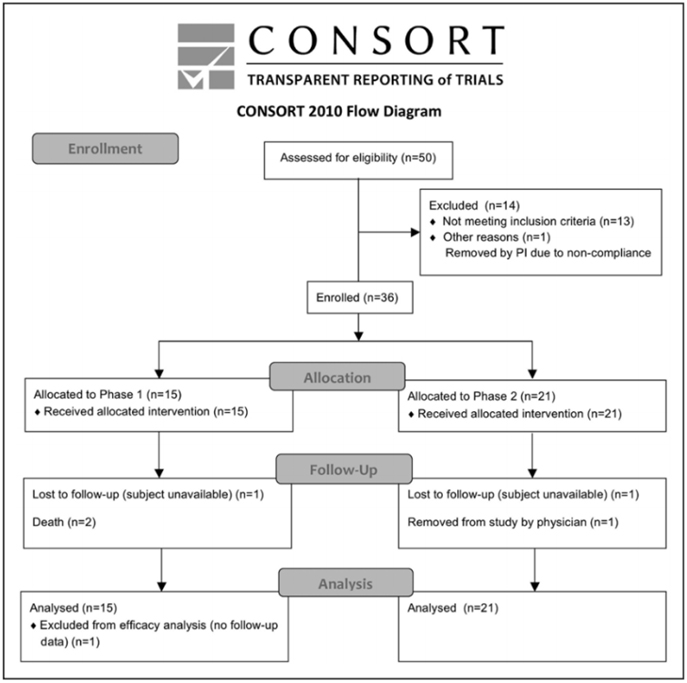
Figure. CONSORT diagram.
Table 2. Baseline Subject Characteristics
| Characteristic | Cohort 1 | Cohort 2 | Cohort 3 | Total |
|---|---|---|---|---|
| n | 5 | 5 | 5 | 21 |
| Sex | ||||
| – Male | 5 (100%) | 4 (80%) | 4 (80%) | 14 (66.67%) |
| – Female | 0 (0%) | 1 (20%) | 1 (20%) | 7 (33.33%) |
| Age, y | 50.8 ± 9.8 | 56.8 ± 11.1 | 68.8 ± 11.58 | 62.8 ± 9.2 |
| [40—62] | [39—69] | [53—84] | [51—83] | |
| Race | ||||
| – White | 4 (80%) | 3 (60%) | 5 (100%) | 17 (80.95%) |
| – Asian | 0 (0%) | 1 (20%) | 0 (0%) | 0 (0%) |
| – American Indian/Alaskan Native | 0 (0%) | 1 (20%) | 0 (0%) | 0 (0%) |
| – Native Hawaiian/Pacific Islander | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| – Black | 1 (20%) | 0 (0%) | 0 (0%) | 1 (4.76%) |
| – Other | 0 (0%) | 0 (0%) | 0 (0%) | 3 (14.29%) |
| Ethnicity | ||||
| – Hispanic or Latino | 0 (0%) | 0 (0%) | 0 (0%) | 2 (9.52%) |
| – Non-Hispanic or Non-Latino | 5 (100%) | 5 (100%) | 5 (100%) | 19 (90.48%) |
| Living situation | ||||
| – At home | 5 (100%) | 5 (100%) | 3 (60%) | 19 (90.48%) |
| – In a living facility | 0 (0%) | 0 (0%) | 2 (40%) | 2 (9.52%) |
| Time from stroke to infusion, y | 1.6 ± 0.9 | 7.7 ± 5.0 | 4.1 ± 2.2 | 4.0 ± 5.0 |
| [0.6—2.9] | [1.1—14.5] | [1.7—7.0] | [0.7—24.8] | |
| Total | 5 | 5 | 5 | 36 |
Safety
A total of 15 serious adverse events were reported. These were wide-ranging in nature, for example, infections, vascular disorders, and pain syndromes (for full details, see Table SIII in the online-only Data Supplement). All serious adverse events were deemed unrelated or unlikely related to the investigational product. A total of 109 adverse events were reported, of which 2, both mild, were considered by the site investigator to be possibly related to the investigational product: one urinary tract infection and one report of intravenous site irritation. Both adverse events recovered completely.
Study testing disclosed no safety concerns. No subject showed a preinfusion positive reaction to intradermal testing. Serial physical exams and blood testing did not disclose any significant findings. Only one of the serial electrocardiograms was thought to have clinically significant findings, in a subject with moderate intraventricular conduction delay, only at the 1-month follow-up visit. Similarly, across serial CT scans of the chest, abdomen, and pelvis, only one was considered clinically significant, a soft tissue density in the anterior abdominal wall seen at 6-months that was stable when reimaged at 12-months.
Behavioral Effects
Across all subjects, improvements were seen in National Institutes of Health Stroke Scale, BI, Mini-Mental Status Exam, and Geriatric Depression Scale scores at both the 6-month and the 12-month follow-up visits (Table 3). These were statistically significant, generally stable over time, and clinically modest in magnitude. Most findings would survive correction for multiple comparisons. Changes in the BI suggest clinical utility, with a 6.8 point gain by 6-months that grew to a 10.8 point gain by 12-months post-infusion (P<0.001), and with the proportion of patients achieving excellent functional outcome (Barthel score =95) increasing from 11.4% (4/35) at baseline to 9/33 (27.3%) at 6-months to 35.5% (11/31) at 12-months.
Discussion
Stroke is a major cause of human disability. This can be reduced by acute therapies that are introduced in the early hours poststroke to reduce initial injury, and by restorative therapies that are introduced days, months, or years poststroke to promote neural repair. Allogeneic MSC show substantial favorable effects in preclinical studies, including when introduced via the intravenous route. The current study found a single intravenous infusion of allogeneic MSC to be safe and potentially associated with functional improvement.
The current study is the largest trial of intravenous MSC in patients with chronic stroke and the first to evaluate allogeneic MSC therapy in this population. It is also the first human stroke study to evaluate MSC grown under hypoxic conditions, which favorably affects cell proliferation, gene expression, cytokine production, and migration. Intravenous infusion of MSC was found to be safe in 36 patients who had chronic stroke with substantial functional deficits. Across 3 escalating doses, treatment-related adverse events were infrequent, mild, and transient. Serial assessments of exam, laboratory testing, electrocardiogram, and CT scans of chest/abdomen/pelvis disclosed no safety concerns, with limited subject dropout. These results are consistent with the overall excellent safety record that MSC have in clinical trials of human subjects across numerous non-cerebrovascular diagnoses and in stroke trials.
Patients with stroke in the chronic stage generally show functional decline; however, enrollees in the current study showed 12 months of continued functional improvement. In general, recovery from stroke-related deficits shows a bimodal time course. Initially, most stroke survivors show some degree of spontaneous recovery, for example, during the initial months for the motor system. Within a year of stroke onset, however, a significant decline in function is commonly seen. This is significant given that few treatment options are available to improve function in patients in the chronic phase of stroke. In the current study, behavioral gains were seen, though were modest in magnitude. However, a 2-point improvement in the National Institutes of Health Stroke Scale score in the setting of chronic stroke, if verified in a larger controlled study, might be regarded as important. Also, the mean gain in BI from baseline grew to 10.8 points by 12 month-poststroke (P<0.001), higher than the BI minimal clinically important difference of 9.25 points. Furthermore, the proportion of patients with an excellent functional outcome (BI score =95) increased from 11.4% at baseline to 27.3% at 6-months and to 35.5% at 12-months. This 12-month period of continued functional improvement is consistent with preclinical studies examining the distribution of systemically administered MSC: intravenous MSC given early after stroke initially localize to lungs then spleen, then increase within the region of brain ischemia, and by 30 days poststroke are concentrated in the peri-infarct region. At one year, most surviving MSC are in the peri-infarct region, with very few present in other organs. Patients also showed significant improvement in the Mini-Mental Status Exam and Geriatric Depression Scale, changes that were largely sustained at 12 months post-infusion, suggesting that MSC have broad effects on brain function. These findings require verification in a larger, controlled study but raise hope that this intervention could improve functional status in the chronic stroke setting. Future studies might also incorporate modality-specific outcome measures to provide more granular assessments of behavioral gains in individual neural systems.
Table 3. Behavioral Change Over Time
| Measure | Baseline | Change to 6 mo | Change to 12 mo |
|---|---|---|---|
| Mini-Mental Status Exam score | 24.2±6.0 (n=35) | 1.8±2.8 (n=32) | 1.3±2.7 (n=31) |
| P Value | <0.001 | 0.017 | |
| NIHSS score | 8 [6.5 to 10] | -1 [-2.25 to 0] | -2 [-3.5 to -0.5] |
| P Value | <0.001 | <0.001 | |
| Geriatric depression scale score | 5.1±3.5 (n=35) | -1.6±3.8 (n=32) | -1.4±3.8 (n=31) |
| P Value | 0.015 | ||
| Barthel Index (score) | 65±28.7 (n=35) | 6.8±11.4 (n=33) | 10.8±15.5 (n=31) |
| P Value | 0.002 | <0.001 | |
| Barthel Index (% =95) | |||
| Proportion at baseline | 4 (11.4%) | 9 (27.3%) | 11 (35.5%) |
| P Value | 0.015 | 0.01 |
Meta-analysis of MSC Effects in Animals with Experimental Ischemic Stroke
Meta-analysis conducted on MSC effects in animals with experimental ischemic stroke demonstrated substantial and sustained effect sizes, even after adjusting for potential publication bias. This effect was consistent across various factors such as species, delivery route, timing of administration post-stroke, and dosage. Preclinical studies have shown that MSC introduction up to 1 month or 4 to 6 weeks post-infarct yields promising results.
Bidirectional Translation: Bedside Experience Informing Preclinical Studies
Findings from patients many months post-stroke (refer to Table 2) underscore the necessity for bidirectional translation. This approach involves translating bedside experiences into insights that inform and refine preclinical studies. This iterative process enhances the applicability and efficacy of future treatments.
Study Strengths
Population with Substantial Functional Deficits
The study enrolled patients in the chronic stage of stroke, a demographic numbering in the millions worldwide, with limited treatment options.
Utilization of Allogeneic MSC
The study utilized allogeneic MSC due to their relatively immunoprivileged nature, eliminating the need for immunosuppression. This approach enables broader implementation within the stroke population.
Safety Evaluation
A dose-escalation design evaluated safety aspects rigorously. Cell culture was limited to 4 passages to optimize MSC features crucial for efficacy.
Study Limitations
Lack of Control Group
Since the study focused primarily on safety, the absence of a control group complicates the interpretation of observed behavioral improvements (refer to Table 3).
Mechanism of Action
The mechanism of action underlying cell therapies improving outcomes in the chronic phase remains unexplored. Future trials should investigate mechanisms like growth factor release, anti-inflammatory effects, and exosome involvement.
Conclusion: Implications for Future Research
This study establishes the safety of intravenous allogeneic MSC in chronic stroke patients with substantial functional deficits. While suggesting functional benefits, further verification through controlled studies is imperative. These findings advocate for continued research into intravenous allogeneic MSC for chronic stroke, including mechanistic exploration.
To improve the on-page SEO for your article and add H2 headings for references, here’s a structured approach:
References
- Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43–53.
- Johnston SC, Hauser SL. Neurological disease on the global agenda. Ann Neurol. 2008;64:A11–A12.
- Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215.
- Lin DJ, Finklestein SP, Cramer SC. New directions in treatments targeting stroke recovery. Stroke. 2018;49:3107–3114. doi: 10.1161/STROKEAHA.118.021359
- Cramer SC. Repairing the human brain after stroke. II. Restorative therapies. Ann Neurol. 2008;63:549–560. doi: 10.1002/ana.21412
- Wolf SL, Winstein CJ, Miller JP, et al; EXCITE Investigators. Effect of constraint-induced movement therapy on upper extremity function 3 to 9 months after stroke: the EXCITE randomized clinical trial. JAMA. 2006;296:2095–2104. doi: 10.1001/jama.296.17.2095
- McCabe J, Monkiewicz M, Holcomb J, et al. Comparison of robotics, functional electrical stimulation, and motor learning methods for treatment of persistent upper extremity dysfunction after stroke: a randomized controlled trial. Arch Phys Med Rehabil. 2015;96:981–990. doi: 10.1016/j.apmr.2014.10.022
- Dodakian L, McKenzie AL, Le V, et al. A home-based telerehabilitation program for patients with stroke. Neurorehabil Neural Repair. 2017;31:923–933. doi: 10.1177/1545968317733818
- Ward NS, Brander F, Kelly K. Intensive upper limb neurorehabilitation in chronic stroke: outcomes from the Queen Square programme. J Neurol Neurosurg Psychiatry. 2019;90:498–506. doi: 10.1136/jnnp-2018-319954
- Vu Q, Xie K, Eckert M, et al. Meta-analysis of preclinical studies of mesenchymal stromal cells for ischemic stroke. Neurology. 2014;82:1277–1286. doi: 10.1212/WNL.0000000000000278
- Bang OY, Lee JS, Lee PH, Lee G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol. 2005;57:874–882. doi: 10.1002/ana.20501
- Honmou O, Houkin K, Matsunaga T, et al. Intravenous administration of auto serum-expanded autologous mesenchymal stem cells in stroke. Brain. 2011;134(pt 6):1790–1807. doi: 10.1093/brain/awr063
- Bhasin A, Srivastava MV, Kumaran SS, et al. Autologous mesenchymal stem cells in chronic stroke. Cerebrovasc Dis Extra. 2011;1:93–104. doi: 10.1159/000333381
- Le Blanc K, Tammik C, Rosendahl K, et al. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896.
- Lalu MM, McIntyre L, Pugliese C, et al; Canadian Critical Care Trials Group. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One. 2012;7:e47559. doi: 10.1371/journal.pone.0047559
- Hess DC, Wechsler LR, Clark WM, et al. Safety and efficacy of multipotent adult progenitor cells in acute ischemic stroke (MASTERS): a randomized, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16:360–368. doi: 10.1016/S1474-4422(17)30046-7
- Steinberg GK, Kondziolka D, Wechsler LR, et al. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a study. Stroke. 2016;47:1817–1824. doi: 10.1161/STROKEAHA.116.012995
- Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078932.pdf. Accessed June 22, 2019.
- Luger D, Lipinski MJ, Westman PC, et al. Intravenously delivered mesenchymal stem cells: systemic anti-inflammatory effects improve left ventricular dysfunction in acute myocardial infarction and ischemic cardiomyopathy. Circ Res. 2017;120:1598–1613. doi: 10.1161/CIRCRESAHA.117.310599
- Harach T, Jammes F, Muller C, et al. Administrations of human adult ischemia-tolerant mesenchymal stem cells and factors reduce amyloid beta pathology in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2017;51:83–96. doi: 10.1016/j.neurobiolaging.2016.11.009
- Butler J, Epstein SE, Greene SJ, et al. Intravenous allogeneic mesenchymal stem cells for nonischemic cardiomyopathy: safety and efficacy results of a phase II-a randomized trial. Circ Res. 2017;120:332–340. doi: 10.1161/CIRCRESAHA.116.309717
- Vertelov G, Kharazi L, Muralidhar MG, et al. High targeted migration of human mesenchymal stem cells grown in hypoxia is associated with enhanced activation of RhoA. Stem Cell Res Ther. 2013;4:5. doi: 10.1186/scrt153
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905
- Devine SM. Mesenchymal stem cells: will they have a role in the clinic? J Cell Biochem Suppl. 2002;38:73–79.
- Hilfiker A, Kasper C, Hass R, Haverich A. Mesenchymal stem cells and progenitor cells in connective tissue engineering and regenerative medicine: is there a future for transplantation? Langenbecks Arch Surg. 2011;396:489–497. doi: 10.1007/s00423-011-0762-
- Published in Corporate News / Blog
Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells

Abstract
Mesenchymal stem/stromal cells (MSCs) are increasingly used therapeutically via intravenous administration due to their efficacy in tissue repair and anti-inflammatory roles. However, comprehensive data on MSC biodistribution, target structures, and migration mechanisms remain sparse. This review explores current hypotheses regarding MSC tissue targeting, covering both preclinical and clinical studies.
Background
In the 1970s, Friedenstein et al. [1] first reported the localized application of culture-expanded bone marrow stroma-derived fibroblastic cells, initiating ectopic hematopoiesis under the kidney capsule. Caplan’s group later defined MSCs as multipotent cells capable of differentiating into various tissues, highlighting their regenerative potential in bone and cartilage [2–4]. Initial studies primarily focused on site-specific effects without addressing biodistribution.
By 2000, interest grew in intravenous MSC application, spurred by Horwitz’s pivotal studies in children with osteogenesis imperfecta [5]. These studies demonstrated MSC efficacy in bone marrow deficiency, suggesting MSC homing to bone sites. Subsequent studies further explored intravenous MSC therapy, employing diverse labeling techniques to track MSC migration across tissues [7–11].
Establishment of Methods to Track Intravenously Administered MSCs
Post-2000, numerous studies in animals and humans utilized various labeling methods to track intravenously administered MSCs. Techniques included radioactive labels, fluorescent dyes, contrast agents, and reporter genes [7–11]. Despite these advancements, tracking methods often only detected short-term MSC homing and did not confirm cell viability. Studies consistently showed initial MSC accumulation in lungs post-injection, followed by distribution to liver and spleen [12].
Human studies, such as those by Koç et al. [13] in mammary carcinoma patients and others in liver cirrhosis patients [14], validated animal findings, confirming MSC presence in blood and major organs. Notably, lung accumulation decreased over time, contrasting with increasing signals in liver and spleen up to 10 days post-administration [14].
To improve the on-page SEO for your article on the expression of cell adhesion molecules by MSCs and their interaction with endothelial cells, I’ll add appropriate H2 headings and ensure the content is structured effectively:
Expression of Cell Adhesion Molecules by MSCs: Basis for Interaction with Endothelial Cells and Tissue-Directed Extravasation
In theory, the interaction of transplanted MSCs with endothelial cells hinges on adhesion molecules present on MSC surfaces and corresponding counter-receptors on endothelial cells. Here’s a breakdown of the current understanding:
Adhesion Molecule Expression by MSCs
Human MSCs (hMSCs) notably exhibit deficiencies in receptor binding to selectins and their ligands. Key findings include:
- Lack of L-selectin expression.
- Non-functional CD44 as an E-selectin ligand [15].
- Binding to P-selectin via a fucosylated ligand, though not P-selectin glycoprotein ligand (PSGL)-1 [16].
- Enzymatic fucosylation of CD44 by Thankamony and Sackstein enhances MSC binding to endothelial E-selectin, facilitating MSC rolling and extravasation into bone marrow sites [17].
Integrin Expression
MSCs express alpha4beta1 (VLA-4) and alpha5beta1 (VLA-5) integrins, critical for their interaction with endothelial cells and tissue-specific homing [15, 16, 18–20].
Chemokine Receptors
MSCs also express chemokine receptors such as CXCR4, which plays a pivotal role in homing and mobilization of hematopoietic cell types [12, 19, 20].
Summary
These insights underscore MSCs’ challenges in expressing and utilizing adhesion receptors for effective extravasation and tissue-specific homing, akin to leukocyte populations.
To enhance the on-page SEO for your article on common themes in MSC biodistribution research, I’ll structure the content with appropriate H2 headings and ensure clarity:
Table 1: Common Themes in MSC Biodistribution Research
| Theme | Targeted Tissues (Possible Mechanism) | References |
|---|---|---|
| Increased homing after intra-arterial delivery compared with intravenous delivery? | Kidney, Joints, Stroke, Other tissues | [30–34] |
| Side effects of intra-arterial versus intravenous delivery? | Incorporation into vessel wall, Obstruction of microvessels, Vascular occlusion | [23, 35, 38, 39] |
| Targeting of vessel wall and vessel-associated tissues? | Lungs, Lymph nodes, Intestine | [47] |
| Targeting of tissues for regeneration | Myocardium, Beta1 integrins, CCL2 (monocytes), Kidney, Gut and liver, Skin, CCL21, JAM-A, Brain, P/E selectin (CD44), CXCR4/flk-1/EPO-R | [18, 30, 44, 48–55, 56–75] |
| Homing to bone marrow | Bone marrow | [76–81] |
| Biodistribution to the immune system | Macrophages, Dendritic cells, T cells, Unknown target cells, Idoleamine desoxygenase, Prostaglandin E2 | [37–43] |
| Elimination mechanisms? | Antibody formation, Phagocytes | [6, 102] |
| Influence of radiation on homing? | Increased in brain, heart, bone marrow, muscles | [43, 82] |
| Homing in malignancies? | Tumor, Mediated by CCL25, Mediated by sodium iodide symporter under the control of RANTES/CCL-5 promoter, Homed MSCs form tumor-associated fibroblasts | [83–92] |
| Formation of microvesicles | Contribution to/be part of MSC biodistribution, Mediated by horizontal transfer of microRNAs | [14, 63, 93–97] |
In many of the earlier studies
In many of the earlier studies, the target sites as well as the molecular mechanisms governing the interactions of MSCs with the local environment after transplantation (e.g., endothelial cells, target tissue), such as adhesion molecules or signaling mechanisms, were either not addressed or were analyzed only to a minor degree. Moreover, MSCs were often evaluated by microscopy, a method relatively prone to artifacts. Many studies also did not quantify the numbers of MSCs in target or other tissues. Likewise, only few studies reported on the size of the identified MSCs. Despite this lack of information, other themes have emerged, especially research on cues that may regulate the biodistribution of systemically applied MSCs; these include first pass tissues, specifically the lungs, inflammation, irradiation, sites of hypoxia or repair, and cancer (Table 1). As a result, concepts have been raised which imply an ability of MSCs to migrate to specific sites—e.g., MSCs as an “injury drugstore” for several acute clinical situations [21, 22].
First-line accumulation of intravenously administered MSCs in the lungs
The first hurdle for intravenously transplanted MSCs is the lung capillary bed. After culture expansion, MSCs are relatively large cells with an estimated average size of around 30 µm in suspension (ranging from 16–53 µm) [23]. Their size may also vary depending on the osmolarity of the culture media, passage number, and/or cell density during seeding as well as general culture conditions (two-dimensional versus three-dimensional culture). In comparison with MSCs, hematopoietic stem cells have a much smaller diameter, ranging from 4–12 µm depending on the subfraction analyzed [24, 25]. Therefore, obstructive events during lung passage are expected after intravenous administration of MSCs. Lee et al. [26] presented a kinetic study of MSCs accumulating in murine lungs in which up to 80 % of injected cells were found in the lungs within a few minutes after injection. Moreover, formation of emboli in lung vessels was noted. The MSC signal (an Alu sequence DNA marker) fell exponentially, with a half-life of about 24 h and practically complete disappearance after 4 days [26]. Barbash and colleagues [10] confirmed the detection of the overall MSC load in the lungs using 99mTc-labeled MSCs in a rat model with induced myocardial infarction. Murine MSCs also showed deleterious effects in mice, including post-injection lethality, which was not the case after administration of hMSCs [27]. Interaction of human or murine MSCs with lung endothelial cells was dependent on the suspension medium in which the transplanted cells were administered [27]. Adhesion of the MSCs to endothelial cells was found to involve the integrin ligand vascular cell adhesion molecule (VCAM)-1. When comparing MSCs with mononuclear cells from bone marrow, neural stem cells and multipotent adult progenitor cells, Fischer et al. [28] found that MSCs showed the highest interaction with lung endothelia, which could be inhibited by pretreatment with anti-CD49d antibody. In a study by Kerkelä et al. [29], adhesion of MSCs to lung tissue (probably endothelial cells) was dependent on the enzyme treatment used during harvesting of confluent MSCs in culture before transplantation; after treatment with pronase, MSCs more readily cleared the lungs and could be found in other tissues compared with trypsinization treatment. Taken together, these data indicate an active role of the adhesion molecules VLA-4/VCAM-1 on MSCs/endothelial cells during interaction of MSCs with lung tissue. It remains to be clarified, however, whether this is a passive or active process. Also, relatively little is known about possible adhesion molecules other than VLA-4/VCAM-1 which may be operative in the interaction of MSCs with endothelial cell surfaces in the lung. This includes the fucosylation of CD44 to HCELL, a highly active E-selectin ligand on MSCs, which is relevant in bone marrow endothelia but seemingly did not affect lung interactions [15].
In summary, presently there is strong evidence that accumulation of MSCs in the lungs is a key determining factor for their biodistribution. The major adhesion molecule involved seems to be VLA-4/VCAM1. Still, it is not clear to what degree the findings in animal studies are quantitatively transferable to humans (Table 1).
Biodistribution of MSCs after intra-arterial versus intravenous administration
Studies comparing intra-arterial and intravenous application of MSCs have demonstrated a major association between intravenous application and retention of MSCs in the lungs, and their increased accumulation in therapeutic target tissues after intra-arterial injection. Walczak et al. [30] in a rat transient ischemia stroke model applied two independent detection methods (magnetic resonance imaging and Doppler flowmetry). They demonstrated that higher cerebral engraftment rates are associated with impeded cerebral blood flow, and that intra-arterial delivery may be advantageous in ischemic stroke to deliver MSCs to the site of injury. Mäkelä et al. [31] compared intra-arterial and intravenous administration of MSCs labeled with 99mTc, and also found that the intra-arterial transplantation route has a positive impact on the biodistribution of bone marrow-derived MSCs (BM-MSCs) to peripheral tissues. They found that intra-arterial transplantation decreased the deposition of BM-MSCs in the lungs and increased uptake in other organs, especially in the liver. In a study looking at human adipose tissue-derived MSCs in SCID mice, Toupet et al. [32] showed that 15 % of intra-arterially injected MSCs accumulate in inflamed joints during the first month, and 1.5 % over a longer term of >6 months, also favoring intra-arterial over intravenous application for, in their case, anti-inflammatory MSCs. Therapeutic effects of MSCs in kidney have been generally achieved after intra-arterial delivery [33, 34]. Although more studies will be needed, these data suggest that the intra-arterial route of administration is effective in avoiding pulmonary entrapment of BM-MSCs, and may thus improve the biodistribution and bioavailability of transplanted MSCs in clinically relevant tissues for, e.g., tissue repair.
Interactions of MSCs with the blood vessel wall: integration into the vessel wall or transmigration?
As described above, the majority of intravenously injected MSCs are generally detected in the lungs, and in no other tissue at comparable numbers even at later time points. Some groups asked whether MSCs may directly target vessels or perivascular tissue and investigated the fate of MSCs in and around blood vessels. These studies followed the cells using intravital microscopy and histologic examination in different tissues after intra-arterial [23, 30, 35] administration. In the cremaster muscle intravital microscopy model, Furlani et al. [23] observed that the microcirculation was disturbed, with some MSCs obstructing small vessels. In addition, pulmonary emboli were found. Toma et al. [35] also observed occlusion of microvessels and entrapment of the injected MSCs. Moreover, they observed stable integration of some transplanted cells into the vessel wall. Cui et al. [36] reported a risk of vascular occlusion in their rat stroke infarction model after intra-arterial injection, pointing to the fact that local intravasal entrapment of MSCs may frequently occur, and MSCs may obstruct the microcirculation. Currently, however, we lack conclusive data that MSCs that are entrapped in capillaries and/or are incorporated into the vessel wall or adjacent to endothelial cells would relocate (i.e., “home”) to their main tissue of origin, pericytes.
Transplanted MSCs interact with cells of the immune system
Transplanted MSCs have been shown to rapidly interact with immune cell types, which are—at least in part—present also in the bloodstream. In a lung sepsis model, Nemeth et al. [37] observed that MSCs co-localize with lung-resident macrophage cells and induce them to produce anti-inflammatory interleukin (IL)-10 via release of prostaglandin E by MSCs as part of their therapeutic effect. Chiesa et al. [38] showed that interstitial dendritic cells (DCs) decrease their physiological migration from skin to lymph nodes rapidly after intravenous administration of MSCs. They describe that MSCs inhibit Toll-like receptor (TLR)-4-induced activation of DCs, which results in the inhibition of cytokine secretion by DCs, downregulation of adhesion molecules involved in the migration of DCs to the lymph nodes, suppression of DC antigen presentation to CD4+ T cells, and cross-presentation to CD8+ T cells. Akiyama et al. [39] demonstrated that both human and murine MSCs can induce immune suppression by attracting and killing autoreactive T cells through FasL, thereby stimulating transforming growth factor beta production by macrophages and generation of regulatory T cells. The interaction has been shown to involve the secretion of MCP-1 by MSCs. The dying T cells in turn activate macrophages to produce transforming growth factor beta, thus stimulating regulatory T cells and promoting immune tolerance. Possibly, the secretion of anti-inflammatory protein TSG-6 by activated MSCs, which
Potential Mechanisms of Elimination of MSCs from the Circulation
Immune Responses and Antibody Formation
The interaction between transplanted MSCs and immune system cells often triggers xenogeneic and allogeneic immune responses. This can lead to the formation of antibodies or T-cell responses against MSCs, making them undetectable upon subsequent administration in patients [6]. Notably, anti-fetal calf serum antibodies have been documented in patients unresponsive to allogeneic MSC applications [6].
Lung Trapping and Systemic Pathways
Despite efforts to trace transplanted MSCs, they are frequently undetectable or only a small fraction is identifiable. This underscores the potential role of the lung as a “first-pass” tissue and suggests that lung trapping might contribute significantly to the elimination of MSCs from the circulation. Moreover, systemic pathways involved in eliminating MSCs in humans likely play a role in their barely detectable long-term engraftment.
Tissue Repair Signaling and MSC Migration
Myocardial Infarction
In myocardial infarction models, MSC migration is facilitated through the VLA-4/VCAM receptor axis. Studies show that pre-treatment of MSCs with TNF-1alpha enhances their migration via VCAM-1, suggesting a role for beta1 integrins in this process [48]. Additionally, chemokine receptors like CXCR4 influence the homing of MSCs to ischemic myocardial tissue [49].
Kidney Damage
Clinical trials and animal studies have highlighted MSCs’ potential in renal disease therapy. Intravenous administration of MSCs has shown promise in reducing rejection rates and improving renal function post-transplantation [56, 57]. Mechanistically, MSCs aid in repairing the glomerular barrier and mitigating tubular injury in various models of kidney damage [60, 61].
Liver Damage
In liver cirrhosis, MSCs have been observed to migrate to damaged areas, although the mechanisms are not fully elucidated. Studies suggest involvement of corticosteroids and the SDF-1/CXCR4 axis in MSC migration during liver fibrosis [65]. Radioimaging studies have shown gradual accumulation of MSCs in the liver and spleen post-infusion [14].
Gut and Skin
MSCs’ presence has been detected in inflammatory bowel disease models, indicating their ability to home to gut tissues affected by inflammation [67]. Similarly, studies on wound repair have shown MSCs’ potential to differentiate into skin cells, though their efficacy varies [44].
Brain
Research in stroke models demonstrates that MSCs migrate to ischemic brain tissue, delivering neurotrophic factors and modulating microglial activity [72, 73]. Chemokine receptors like CXCR4 play a role in MSC recruitment to inflamed brain areas [74].
Homing of MSCs to Bone Marrow
Engraftment of MSCs in bone marrow depends on specific conditions, such as niche availability. While prolonged culture may impair MSC engraftment in classic bone marrow transplantation, certain deficiencies in recipients may facilitate MSC incorporation [76–80]. Studies indicate that MSCs can mediate stromal engraftment in deficient hosts, contributing to hematopoietic environments [81].
To improve the on-page SEO for the article, I’ve added relevant H2 headings and made adjustments for clarity and structure. Here are the changes:
Potential Mechanisms of Elimination of MSCs from the Circulation
A critical aspect of MSC transplantation involves their interaction with the immune system, both in animal models and humans. Transplanted MSCs can induce xenogeneic and allogeneic immune responses, leading to antibody formation or T-cell responses against them [6]. This immune reaction often results in the failure to detect transplanted MSCs upon repeated administration, particularly when cultured in media containing fetal bovine serum [6]. Studies indicate that anti-fetal calf serum antibodies can develop in patients who do not respond to repeated MSC applications [6]. The elimination mechanisms of xenogeneic MSCs in animals parallel those seen in allogeneic settings.
Despite the identification of several target tissues for MSCs, little data exist on their final destinations or the routes of their elimination after systemic administration. The inability to detect transplanted MSCs or their minimal presence suggests the lung might act as a primary tissue for initial MSC trapping and possibly plays a role in their elimination. Conversely, the systemic pathways responsible for eliminating transplanted MSCs in humans may lead to their barely detectable long-term engraftment.
Influence of Irradiation on Migration and Biodistribution of MSCs
In a murine study, Francois et al. [43] demonstrated that both total body irradiation and local irradiation significantly alter the distribution of intravenously infused hMSCs in NOD/SCID mice compared to untreated controls. Non-irradiated control animals showed minimal presence of infused hMSCs, predominantly in the lung, bone marrow, and muscles. However, mice subjected to total body irradiation exhibited increased absolute numbers of hMSCs in the brain, heart, bone marrow, and muscles. Moreover, selective irradiation of limbs or the abdomen resulted in enhanced engraftment of hMSCs in the exposed skin or muscles compared to total body irradiation alone, indicating both local and systemic effects on MSC engraftment. The study did not investigate long-term engraftment effects.
Sémont et al. [82] investigated the engraftment and efficacy of transplanted MSCs in an immunodeficient mouse model of radiation-induced gastrointestinal tract failure. They observed accelerated recovery in mice receiving hMSCs, characterized by decreased apoptosis of epithelial cells and increased proliferation in the small intestinal mucosa. Despite these benefits, significant amounts of transplanted MSCs were not detected.
A Special Case: Migration and Engraftment of MSCs into Tumors
Tumor-associated fibroblasts, derived from the MSC pool, are integral components of the microenvironment in various solid tumors [83, 84]. Tumor tissue represents a target for intravenously injected MSCs due to their tumor tropism. Experimental studies have reported both beneficial and adverse effects of MSC migration into tumor areas. For instance, Beckermann et al. [85] observed migration of MSCs into areas adjacent to vessel walls in human pancreatic tumors in immunodeficient mice. Additionally, Alieva et al. [86] tracked adipose tissue-derived MSCs in a glioblastoma model, demonstrating their incorporation and subsequent elimination upon gancyclovir administration, leading to tumor regression. These studies underscore the potential utility of MSCs in targeting tumors using approaches like suicide transgene therapy.
Recent Developments: Exosomes, Microparticles, and MSCs
MSCs have the capacity to produce exosomes, small membrane vesicles that play critical roles in mediating therapeutic effects. Exosomes derived from MSCs have been implicated in various target tissues, such as tubular cells in acute kidney injury and post-traumatic brain injury recovery [63, 93, 94]. These vesicles contain signaling molecules that facilitate MSC-mediated therapeutic effects through horizontal transfer mechanisms. Recent studies, such as Kordelas et al. [98], have demonstrated the therapeutic potential of MSC-derived exosomes in treating severe graft-versus-host disease, highlighting their expanding role in regenerative medicine.
Summary: Possible Interactions of MSCs within the Circulatory System and Their Biodistribution
MSCs interact dynamically within the bloodstream, influencing their biodistribution and interactions with the immune system. These interactions include modulation of T-cell responses, evasion of NK cell cytoxicity, and potential triggers of inflammatory responses upon their introduction into the bloodstream [46, 99, 102]. Understanding these interactions is crucial for optimizing MSC-based therapies and enhancing their therapeutic efficacy in various clinical settings.
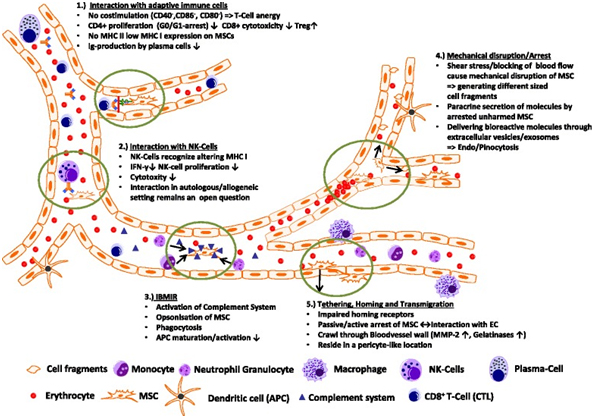
Conclusion
The fate of intravenously injected MSCs remains a puzzle, as evidenced by limited detection in preclinical animal studies and human trials. Numerous questions persist, such as the nature of interactions between MSCs and host cells upon infusion into the bloodstream, the clearance pathways for non-migrating MSCs, and the relevance of intact MSCs in observed therapeutic effects.
Further comprehensive analysis using animal disease models is essential to elucidate the roles of mediators like exosomes, signaling proteins, and microRNAs. These investigations will advance our understanding of MSC biodistribution, migration, homing mechanisms, and the underlying mechanisms of their therapeutic benefits. Insights gained from these studies hold promise for refining MSC-derived therapies and developing new therapeutic strategies.
References
- Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal cells responsible for transferring the micro-environment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. 1974;17:331–40. doi: 10.1097/00007890-197404000-00001. [PubMed] [CrossRef] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [PubMed] [CrossRef] [Google Scholar]
- Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–84. doi: 10.1002/jcb.20886. [PubMed] [CrossRef] [Google Scholar]
- Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217:318–24. doi: 10.1002/path.2469. [PubMed] [CrossRef] [Google Scholar]
- Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–7. doi: 10.1073/pnas.132252399. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–13. doi: 10.1038/6529. [PubMed] [CrossRef] [Google Scholar]
- Devine SM, Bartholomew A, Mahmud N, Nelson M, Patil S, Hardy W, et al. Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol. 2001;29:244–55. doi: 10.1016/S0301-472X(00)00635-4. [PubMed] [CrossRef] [Google Scholar]
- Devine SM, Cobbs C, Jennings M, Bartholomew A, Hoffman R. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003;101:2999–3001. doi: 10.1182/blood-2002-06-1830. [PubMed] [CrossRef] [Google Scholar]
- Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169:12–20. doi: 10.1159/000047856. [PubMed] [CrossRef] [Google Scholar]
- Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863–8. doi: 10.1161/01.CIR.0000084828.50310.6A. [PubMed] [CrossRef] [Google Scholar]
- Kraitchman D, Tatsumi M, Gilson WD, Ishimori T, Kedziorek D, Walczak P, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–61. doi: 10.1161/CIRCULATIONAHA.105.537480. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–16. doi: 10.1016/j.stem.2009.02.001. [PubMed] [CrossRef] [Google Scholar]
- Koç ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–16. [PubMed] [Google Scholar]
- Gholamrezanezhad A, Mirpour S, Bagheri M, Mohamadnejad M, Alimoghaddam K, Abdolahzadeh L, et al. In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol. 2011;38:961–7. doi: 10.1016/j.nucmedbio.2011.03.008. [PubMed] [CrossRef] [Google Scholar]
- Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–7. doi: 10.1038/nm1703. [PubMed] [CrossRef] [Google Scholar]
- Ruster B, Gottig S, Ludwig RJ, Bistrian R, Muller S, Seifried E, et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108:3938–44. doi: 10.1182/blood-2006-05-025098. [PubMed] [CrossRef] [Google Scholar]
- Thankamony SP, Sackstein R. Enforced hematopoietic cell E- and L-selectin ligand (HCELL) expression primes transendothelial migration of human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2011;108:2258–63. doi: 10.1073/pnas.1018064108. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, et al. Monocyte chemotactic protein-3 is a myocardial MSC homing factor. Stem Cells. 2007;25:245–51. doi: 10.1634/stemcells.2006-0293. [PubMed] [CrossRef] [Google Scholar]
- Shi M, Li J, Liao L, Chen B, Li B, Chen L, et al. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007;92:897–904. doi: 10.3324/haematol.10669. [PubMed] [CrossRef] [Google Scholar]
- Potapova IA, Brink PR, Cohen IS, Doronin SV. Culturing of human mesenchymal stem cells as three-dimensional aggregates induces functional expression of CXCR4 that regulates adhesion to endothelial cells. J Biol Chem. 2008;283:13100–7. doi: 10.1074/jbc.M800184200. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Caplan A, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11–5. doi: 10.1016/j.stem.2011.06.008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324:1666–9. doi: 10.1126/science.1172687. [PubMed] [CrossRef] [Google Scholar]
- Furlani D, Ugurlucan M, Ong L, Bieback K, Pittermann E, Westien I, et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc Res. 2009;77:370–6. doi: 10.1016/j.mvr.2009.02.001. [PubMed] [CrossRef] [Google Scholar]
- Sharma S, Krishan A. Hematopoietic Stem Cells. In: Krishan A, Krishnamurthy H, Totey S, editors. Applications of flow cytometry in stem cell research and tissue regeneration. New Jersey: Wiley-Blackwell; 2010. pp. 103–14. [Google Scholar]
- Radley JM, Ellis S, Palatsides M, Williams B, Bertoncello I. Ultrastructure of primitive hematopoietic stem cells isolated using probes of functional status. Exp Hematol. 1999;27:365–9. doi: 10.1016/S0301-472X(98)00017-4. [PubMed] [CrossRef] [Google Scholar]
- Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Deak E, Rüster B, Keller L, Eckert K, Fichtner I, Seifried E, et al. Suspension medium influences interaction of mesenchymal stromal cells with endothelium and pulmonary toxicity after transplantation in mice. Cytotherapy. 2010;12:260–4. doi: 10.3109/14653240903401840. [PubMed] [CrossRef] [Google Scholar]
- Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18(5):683–92. doi: 10.1089/scd.2008.0253. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kerkelä E, Hakkarainen T, Mäkelä T, Raki M, Kambur O, Kilpinen L, et al. Transient proteolytic modification of mesenchymal stromal cells increases lung clearance rate and targeting to injured tissue. Stem Cells Transl Med. 2013;2(7):510–20. doi: 10.5966/sctm.2012-0187. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Walczak P, Zhang J, Gilad AA, Kedziorek DA, Ruiz-Cabello J, Young RG, et al. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke. 2008;39:1569–74. doi: 10.1161/STROKEAHA.107.502047. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Mäkelä T, Takalo R, Arvola O, Haapanen H, Yannopoulos F, Blanco R, et al. Safety and biodistribution study of bone marrow-derived mesenchymal stromal cells and mononuclear cells and the impact of the administration route in an intact porcine model. Cytotherapy. 2015;17:392–402. doi: 10.1016/j.jcyt.2014.12.004. [PubMed] [CrossRef] [Google Scholar]
- Toupet K, Maumus M, Luz-Crawford P, Lombardo E, Lopez-Belmonte J, van Lent P, et al. Survival and biodistribution of xenogenic adipose mesenchymal stem cells is not affected by the degree of inflammation in arthritis. PLoS One. 2015;10:e0114962. doi: 10.1371/journal.pone.0114962. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tögel F, Yang Y, Zhang P, Hu Z, Westenfelder C. Bioluminescence imaging to monitor the in vivo distribution of administered mesenchymal stem cells in acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F315–21. doi: 10.1152/ajprenal.00098.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Morigi M, De Coppi P. Cell therapy for kidney injury: different options and mechanisms—mesenchymal and amniotic fluid stem cells. Nephron Exp Nephrol. 2014;126:59. doi: 10.1159/000360667. [PubMed] [CrossRef] [Google Scholar]
- Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104(3):398–402. doi: 10.1161/CIRCRESAHA.108.187724. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Cui LL, Kerkelä E, Bakreen A, Nitzsche F, Andrzejewska A, Nowakowski A, et al. The cerebral embolism evoked by intraaterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cell Res Ther. 2015;6:11. doi: 10.1186/scrt544. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–9. doi: 10.1038/nm.1905. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Chiesa S, Morbelli S, Morando S, Massollo M, Marini C, Bertoni A, et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. Proc Natl Acad Sci U S A. 2011;108:17384–9. doi: 10.1073/pnas.1103650108. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, et al. Mesenchymal stem cell-induced immunoregulation involves Fas ligand/Fas-mediated T cell apoptosis. Cell Stem Cell. 2012;10(5):544–55. doi: 10.1016/j.stem.2012.03.007. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-?B signaling in resident macrophages. Blood. 2011;118:330–8. doi: 10.1182/blood-2010-12-327353. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kim J, Hematti P. Mesenchymal stem celleducated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37:1445–53. doi: 10.1016/j.exphem.2009.09.004. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One. 2010;5(2):e9252. doi: 10.1371/journal.pone.0009252. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Francois M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2011;20:187–95. doi: 10.1038/mt.2011.189. [PubMed] [CrossRef] [Google Scholar]
- Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180:2581–7. doi: 10.4049/jimmunol.180.4.2581. [PubMed] [CrossRef] [Google Scholar]
- Le Blanc K, Mougiakanos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. 2012;12:383–96. doi: 10.1038/nri3209. [PubMed] [CrossRef] [Google Scholar]
- Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–506. doi: 10.1182/blood-2007-02-069716. [PubMed] [CrossRef] [Google Scholar]
- von Bahr L, Batsis I, Moll G, Hägg M, Szakos A, Sundberg B, et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575–8. doi: 10.1002/stem.1118. [PubMed] [CrossRef] [Google Scholar]
- Segers VFM, Van Riet I, Andries LJ, Lemmens K, Demolder MJ, De Becker AJML, et al. Mesenchymal stem cell adhesion to cardiac microvascular endothelium: activators and mechanisms. Am J Physiol Heart Circ Physiol. 2006;290:H1370–7. doi: 10.1152/ajpheart.00523.2005. [PubMed] [CrossRef] [Google Scholar]
- Ip JE, Wu Y, Huang J, Zhang L, Pratt RE, Dzau VJ. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol Biol Cell. 2007;18:2873–82. doi: 10.1091/mbc.E07-02-0166. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu Y, Zhao RC. The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012;8:243–50. doi: 10.1007/s12015-011-9293-z. [PubMed] [CrossRef] [Google Scholar]
- Zhang M, Mal N, Kiedrowski M, Chacko M, Askari AT, Popovic ZB, et al. SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. FASEB J. 2007;21:3197–207. doi: 10.1096/fj.06-6558com. [PubMed] [CrossRef] [Google Scholar]
- Belema-Bedada F, Uchida S, Martire A, Kostin S, Braun T. Efficient homing of multipotent adult MSCs depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell. 2008;2:566–75. doi: 10.1016/j.stem.2008.03.003. [PubMed] [CrossRef] [Google Scholar]
- Wang Y, Zhang D, Ashraf M, Zhao T, Huang W, Ashraf A, et al. Combining neuropeptide Y and mesenchymal stem cells reverses remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;298:H275–86. doi: 10.1152/ajpheart.00765.2009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Laurila JP, Laatikainen L, Castellone MD, Trivedi P, Heikkila J, Hinkkanen A, et al. Human embryonic stem cell-derived mesenchymal stromal cell transplantation in a rat hind limb injury model. Cytotherapy. 2009;11:726–37. doi: 10.3109/14653240903067299. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Jasmin, Jelicks LA, Tanowitz HB, Peters VM, Mendez-Otero R, de Carvalho AC C, et al. Molecular imaging, biodistribution and efficacy of mesenchymal bone marrow cell therapy in a mouse model of Chagas disease. Microbes Infect. 2014;16:923–35. doi: 10.1016/j.micinf.2014.08.016. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Reinders ME, de Fijter JW, Roelofs H, Bajema IM, de Vries DK, Schaapherder AF, et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl Med. 2013;2:107–11. doi: 10.5966/sctm.2012-0114. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. 2012;307:1169–77. doi: 10.1001/jama.2012.316. [PubMed] [CrossRef] [Google Scholar]
- Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–25. doi: 10.1146/annurev.med.59.061506.154239. [PubMed] [CrossRef] [Google Scholar]
- Gooch A, Doty J, Flores J, Swenson L, Toegel FE, Reiss GR, et al. Initial report on a phase I clinical trial: prevention and treatment of post-operative acute kidney injury with allogeneic mesenchymal stem cells in patients who require on-pump cardiac surgery. Cell Ther Transplant. 2008;1:e.000028.01. [Google Scholar]
- Sugimoto H, Mundel TM, Sund M. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci U S A. 2006;103:7321–6. doi: 10.1073/pnas.0601436103. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Morigi M, Imberti B, Zoja C, Corna D, Tomasoni S, Abbate M, et al. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J Am Soc Nephrol. 2004;15(7):1794–804. doi: 10.1097/01.ASN.0000128974.07460.34. [PubMed] [CrossRef] [Google Scholar]
- Imberti B, Morigi M, Tomasoni S, Rota C, Corna D, Longaretti L, et al. Insulin-like growth factor-1 sustains stem cell mediated renal repair. J Am Soc Nephrol. 2007;18:2921–8. doi: 10.1681/ASN.2006121318. [PubMed] [CrossRef] [Google Scholar]
- Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–67. doi: 10.1681/ASN.2008070798. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Briquet A, Grégoire C, Comblain F, Servais L, Zeddou M, Lechanteur C, et al. Human bone marrow, umbilical cord or liver mesenchymal stromal cells fail to improve liver function in a model of CCl4-induced liver damage in NOD/SCID/IL-2R?(null) mice. Cytotherapy. 2014;16:1511–8. doi: 10.1016/j.jcyt.2014.07.003. [PubMed] [CrossRef] [Google Scholar]
- Zhang S, Lv C, Yang X, Han Z, Zhang S, Zhang J, et al. Corticosterone mediates the inhibitory effect of restraint stress on the migration of mesenchymal stem cell to carbon tetrachloride-induced fibrotic liver by downregulating CXCR4/7 expression. Stem Cells Dev. 2015;24(5):587–96. doi: 10.1089/scd.2014.0243. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Li Q, Zhou X, Shi Y, Li J, Zheng L, Cui L, et al. In vivo tracking and comparison of the therapeutic effects of MSCs and HSCs for liver injury. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0062363. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Parekkadan B, Upadhyay R, Dunham J, Iwamoto Y, Mizoguchi E, Mizoguchi A, et al. Bone marrow stromal cell transplants prevent experimental enterocolitis and require host CD11b?+?splenocytes. Gastroenterology. 2011;140:966–75. doi: 10.1053/j.gastro.2010.10.013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu M, Ji S, Xiao S, Kong Z, Fang H, Zhang Y, et al. JAM-A promotes wound healing by enhancing both homing and secretion activities of mesenchymal stem cells. Clin Sci (Lond). 2015. [Epub ahead of print]. [PubMed]
- Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614. doi: 10.1002/stem.187. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Fathke C, Wilson L, Hutter J, Kapoor V, Smith A, Hocking A, et al. Contribution of bone marrow-derived cells to skin: collagen deposition and wound repair. Stem Cells. 2004;22:812. doi: 10.1634/stemcells.22-5-812. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu Y, Chen L, Scott PG, Tredget EE. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells. 2007;25:2648. doi: 10.1634/stemcells.2007-0226. [PubMed] [CrossRef] [Google Scholar]
- Wu J, Sun Z, Sun H-S, Wu J, Weisel RD, Keating A, et al. Intravenously administered bone marrow cells migrate to damaged brain tissue and improve neural function in ischemic rats. Cell Transplant. 2008;16:993–1005. doi: 10.3727/000000007783472435. [PubMed] [CrossRef] [Google Scholar]
- Yilmaz G, Vital S, Yilmaz CE, Stokes KY, Alexander JS, Granger DN. Selectin-mediated recruitment of bone marrow stromal cells in the postischemic cerebral microvasculature. Stroke. 2011;42:806–11. doi: 10.1161/STROKEAHA.110.597088. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wei L, Fraser JL, Lu ZY, Hu X, Yu SP. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol Dis. 2012;46:635–45. doi: 10.1016/j.nbd.2012.03.002. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Constantin G, Marconi S, Rossi B, Angiari S, Calderan L, Anghileri E, et al. Adipose-derived mesenchymal stem cells ameliorate chronic experimental autoimmune encephalomyelitis. Stem Cells. 2009;27:2624–35. doi: 10.1002/stem.194. [PubMed] [CrossRef] [Google Scholar]
- Simmons PJ, Przepiorka D, Thomas ED, Torok-Storb B. Host origin of marrow stromal cells following allogeneic bone marrow transplantation. Nature. 1987;328:429–32. doi: 10.1038/328429a0. [PubMed] [CrossRef] [Google Scholar]
- Cilloni D, Carlo-Stella C, Falzetti F, Sammarelli G, Regazzi E, Colla S, et al. Limited engraftment capacity of bone marrow-derived mesenchymal cells following T-cell-depleted hematopoietic stem cell transplantation. Blood. 2000;96:3637–43. [PubMed] [Google Scholar]
- Rieger K, Marinets O, Fietz T, Körper S, Sommer D, Mücke C, et al. Mesenchymal stem cells remain of host origin even a long time after allogeneic peripheral blood stem cell or bone marrow transplantation. Exp Hematol. 2005;33:605–11. doi: 10.1016/j.exphem.2005.02.004. [PubMed] [CrossRef] [Google Scholar]
- Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–70. doi: 10.1038/sj.leu.2402763. [PubMed] [CrossRef] [Google Scholar]
- Follenzi A, Raut S, Merlin S, Sarkar R, Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119:5532–42. doi: 10.1182/blood-2011-07-367680. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Otsuru S, Gordon PL, Shimono K, Jethva R, Marino R, Phillips CL, et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood. 2012;120:1933–41. doi: 10.1182/blood-2011-12-400085. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Sémont A, Mouiseddine M, François A, Demarquay C, Mathieu N, Chapel A, et al. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ. 2010;17:952–61. doi: 10.1038/cdd.2009.187. [PubMed] [CrossRef] [Google Scholar]
- Fakhrejahani E, Toi M. Tumor angiogenesis: pericytes and maturation are not to be ignored. J Oncol. 2012;2012:261750. doi: 10.1155/2012/261750. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- D’souza N, Burns JS, Grisendi G, Candini O, Veronesi E, Piccinno S, et al. MSC and tumors: homing, differentiation, and secretion influence therapeutic potential. Adv Biochem Eng Biotechnol. 2013;130:209–66. [PubMed] [Google Scholar]
- Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008;99:622–31. doi: 10.1038/sj.bjc.6604508. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Alieva M, Bagó JR, Aguilar E, Soler-Botija C, Vila OF, Molet J, et al. Glioblastoma therapy with cytotoxic mesenchymal stromal cells optimized by bioluminescence imaging of tumor and therapeutic cell response. PLoS One. 2012;7:e35148. doi: 10.1371/journal.pone.0035148. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Knoop K, Schwenk N, Schmohl K, Müller A, Zach C, Cyran C, et al. Mesenchymal stem cell (MSC)-mediated, tumor stroma-targeted radioiodine therapy of metastatic colon cancer using the sodium iodide symporter as theranostic gene. J Nucl Med. 2015;pii:jnumed.114.146662. [PubMed]
- Xu S, Menu E, De Becker A, Van Camp B, Vanderkerken K, Van Riet I. Bone marrow-derived mesenchymal stromal cells are attracted by multiple myeloma cell-produced chemokine CCL25 and favor myeloma cell growth in vitro and in vivo. Stem Cells. 2012;30:266–79. doi: 10.1002/stem.787. [PubMed] [CrossRef] [Google Scholar]
- Duan X, Guan H, Cao Y, Kleinerman ES. Murine bone marrow-derived mesenchymal stem cells as vehicles for interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer. 2009;115:13–22. doi: 10.1002/cncr.24013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Grisendi G, Bussolari R, Veronesi E, Piccinno S, Burns JS, De Santis G, et al. Understanding tumor-stroma interplays for targeted therapies by armed mesenchymal stromal progenitors: the Mesenkillers. Am J Cancer Res. 2011;1:787–805. [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J, et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med. 2011;17:579–87. doi: 10.2119/molmed.2010.00157. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–57. doi: 10.3390/ijms15034142. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Akyurekli C, Le Y, Richardson RB, Fergusson D, Tay J, Allan DS. A systematic review of preclinical studies on the therapeutic potential of mesenchymal stromal cell-derived microvesicles. Stem Cell Rev. 2015;11:150–60. doi: 10.1007/s12015-014-9545-9. [PubMed] [CrossRef] [Google Scholar]
- Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, et al. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J Neurosurg. 2015;16:1–12. [PMC free article] [PubMed] [Google Scholar]
- Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y, et al. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. 2013;31:2737–46. doi: 10.1002/stem.1409. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tomasoni S, Longaretti L, Rota C, Morigi M, Conti S, Gotti E, et al. Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev. 2013;22:772–80. doi: 10.1089/scd.2012.0266. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kordelas L, Rebmann V, Ludwig AK, Radtke S, Ruesing J, Doeppner TR, et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970–3. [PubMed] [Google Scholar]
- Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. 2006;107:1484–90. doi: 10.1182/blood-2005-07-2775. [PubMed] [CrossRef] [Google Scholar]
- Giuliani M, Bennaceur-Griscelli A, Nanbakhsk A, Oudrhiri N, Chouaib S, Azzarone B, et al. TLR ligands stimulation protects MSC from NK killing. Stem Cells. 2014;32:290–300. doi: 10.1002/stem.1563. [PubMed] [CrossRef] [Google Scholar]
- Reinders ME, Hoogduijn MJ. NK cells and MSCs: possible implications for MSC therapy in renal transplantation. J Stem Cell Res Ther. 2014;4:10000166. doi: 10.4172/2157-7633.1000166. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Moll G, Rasmusson-Duprez I, Von Bahr L, Conolly-Andersen AM, Elgue G, Funke L, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood. Stem Cells. 2012;30:1565–74. doi: 10.1002/stem.1111. [PubMed] [CrossRef] [Google Scholar]
Articles from Stem Cell Research & Therapy are provided here courtesy of BioMed Central
- Published in Corporate News / Blog
