Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells

Abstract
Mesenchymal stem/stromal cells (MSCs) are increasingly used therapeutically via intravenous administration due to their efficacy in tissue repair and anti-inflammatory roles. However, comprehensive data on MSC biodistribution, target structures, and migration mechanisms remain sparse. This review explores current hypotheses regarding MSC tissue targeting, covering both preclinical and clinical studies.
Background
In the 1970s, Friedenstein et al. [1] first reported the localized application of culture-expanded bone marrow stroma-derived fibroblastic cells, initiating ectopic hematopoiesis under the kidney capsule. Caplan’s group later defined MSCs as multipotent cells capable of differentiating into various tissues, highlighting their regenerative potential in bone and cartilage [2–4]. Initial studies primarily focused on site-specific effects without addressing biodistribution.
By 2000, interest grew in intravenous MSC application, spurred by Horwitz’s pivotal studies in children with osteogenesis imperfecta [5]. These studies demonstrated MSC efficacy in bone marrow deficiency, suggesting MSC homing to bone sites. Subsequent studies further explored intravenous MSC therapy, employing diverse labeling techniques to track MSC migration across tissues [7–11].
Establishment of Methods to Track Intravenously Administered MSCs
Post-2000, numerous studies in animals and humans utilized various labeling methods to track intravenously administered MSCs. Techniques included radioactive labels, fluorescent dyes, contrast agents, and reporter genes [7–11]. Despite these advancements, tracking methods often only detected short-term MSC homing and did not confirm cell viability. Studies consistently showed initial MSC accumulation in lungs post-injection, followed by distribution to liver and spleen [12].
Human studies, such as those by Koç et al. [13] in mammary carcinoma patients and others in liver cirrhosis patients [14], validated animal findings, confirming MSC presence in blood and major organs. Notably, lung accumulation decreased over time, contrasting with increasing signals in liver and spleen up to 10 days post-administration [14].
To improve the on-page SEO for your article on the expression of cell adhesion molecules by MSCs and their interaction with endothelial cells, I’ll add appropriate H2 headings and ensure the content is structured effectively:
Expression of Cell Adhesion Molecules by MSCs: Basis for Interaction with Endothelial Cells and Tissue-Directed Extravasation
In theory, the interaction of transplanted MSCs with endothelial cells hinges on adhesion molecules present on MSC surfaces and corresponding counter-receptors on endothelial cells. Here’s a breakdown of the current understanding:
Adhesion Molecule Expression by MSCs
Human MSCs (hMSCs) notably exhibit deficiencies in receptor binding to selectins and their ligands. Key findings include:
- Lack of L-selectin expression.
- Non-functional CD44 as an E-selectin ligand [15].
- Binding to P-selectin via a fucosylated ligand, though not P-selectin glycoprotein ligand (PSGL)-1 [16].
- Enzymatic fucosylation of CD44 by Thankamony and Sackstein enhances MSC binding to endothelial E-selectin, facilitating MSC rolling and extravasation into bone marrow sites [17].
Integrin Expression
MSCs express alpha4beta1 (VLA-4) and alpha5beta1 (VLA-5) integrins, critical for their interaction with endothelial cells and tissue-specific homing [15, 16, 18–20].
Chemokine Receptors
MSCs also express chemokine receptors such as CXCR4, which plays a pivotal role in homing and mobilization of hematopoietic cell types [12, 19, 20].
Summary
These insights underscore MSCs’ challenges in expressing and utilizing adhesion receptors for effective extravasation and tissue-specific homing, akin to leukocyte populations.
To enhance the on-page SEO for your article on common themes in MSC biodistribution research, I’ll structure the content with appropriate H2 headings and ensure clarity:
Table 1: Common Themes in MSC Biodistribution Research
| Theme | Targeted Tissues (Possible Mechanism) | References |
|---|---|---|
| Increased homing after intra-arterial delivery compared with intravenous delivery? | Kidney, Joints, Stroke, Other tissues | [30–34] |
| Side effects of intra-arterial versus intravenous delivery? | Incorporation into vessel wall, Obstruction of microvessels, Vascular occlusion | [23, 35, 38, 39] |
| Targeting of vessel wall and vessel-associated tissues? | Lungs, Lymph nodes, Intestine | [47] |
| Targeting of tissues for regeneration | Myocardium, Beta1 integrins, CCL2 (monocytes), Kidney, Gut and liver, Skin, CCL21, JAM-A, Brain, P/E selectin (CD44), CXCR4/flk-1/EPO-R | [18, 30, 44, 48–55, 56–75] |
| Homing to bone marrow | Bone marrow | [76–81] |
| Biodistribution to the immune system | Macrophages, Dendritic cells, T cells, Unknown target cells, Idoleamine desoxygenase, Prostaglandin E2 | [37–43] |
| Elimination mechanisms? | Antibody formation, Phagocytes | [6, 102] |
| Influence of radiation on homing? | Increased in brain, heart, bone marrow, muscles | [43, 82] |
| Homing in malignancies? | Tumor, Mediated by CCL25, Mediated by sodium iodide symporter under the control of RANTES/CCL-5 promoter, Homed MSCs form tumor-associated fibroblasts | [83–92] |
| Formation of microvesicles | Contribution to/be part of MSC biodistribution, Mediated by horizontal transfer of microRNAs | [14, 63, 93–97] |
In many of the earlier studies
In many of the earlier studies, the target sites as well as the molecular mechanisms governing the interactions of MSCs with the local environment after transplantation (e.g., endothelial cells, target tissue), such as adhesion molecules or signaling mechanisms, were either not addressed or were analyzed only to a minor degree. Moreover, MSCs were often evaluated by microscopy, a method relatively prone to artifacts. Many studies also did not quantify the numbers of MSCs in target or other tissues. Likewise, only few studies reported on the size of the identified MSCs. Despite this lack of information, other themes have emerged, especially research on cues that may regulate the biodistribution of systemically applied MSCs; these include first pass tissues, specifically the lungs, inflammation, irradiation, sites of hypoxia or repair, and cancer (Table 1). As a result, concepts have been raised which imply an ability of MSCs to migrate to specific sites—e.g., MSCs as an “injury drugstore” for several acute clinical situations [21, 22].
First-line accumulation of intravenously administered MSCs in the lungs
The first hurdle for intravenously transplanted MSCs is the lung capillary bed. After culture expansion, MSCs are relatively large cells with an estimated average size of around 30 µm in suspension (ranging from 16–53 µm) [23]. Their size may also vary depending on the osmolarity of the culture media, passage number, and/or cell density during seeding as well as general culture conditions (two-dimensional versus three-dimensional culture). In comparison with MSCs, hematopoietic stem cells have a much smaller diameter, ranging from 4–12 µm depending on the subfraction analyzed [24, 25]. Therefore, obstructive events during lung passage are expected after intravenous administration of MSCs. Lee et al. [26] presented a kinetic study of MSCs accumulating in murine lungs in which up to 80 % of injected cells were found in the lungs within a few minutes after injection. Moreover, formation of emboli in lung vessels was noted. The MSC signal (an Alu sequence DNA marker) fell exponentially, with a half-life of about 24 h and practically complete disappearance after 4 days [26]. Barbash and colleagues [10] confirmed the detection of the overall MSC load in the lungs using 99mTc-labeled MSCs in a rat model with induced myocardial infarction. Murine MSCs also showed deleterious effects in mice, including post-injection lethality, which was not the case after administration of hMSCs [27]. Interaction of human or murine MSCs with lung endothelial cells was dependent on the suspension medium in which the transplanted cells were administered [27]. Adhesion of the MSCs to endothelial cells was found to involve the integrin ligand vascular cell adhesion molecule (VCAM)-1. When comparing MSCs with mononuclear cells from bone marrow, neural stem cells and multipotent adult progenitor cells, Fischer et al. [28] found that MSCs showed the highest interaction with lung endothelia, which could be inhibited by pretreatment with anti-CD49d antibody. In a study by Kerkelä et al. [29], adhesion of MSCs to lung tissue (probably endothelial cells) was dependent on the enzyme treatment used during harvesting of confluent MSCs in culture before transplantation; after treatment with pronase, MSCs more readily cleared the lungs and could be found in other tissues compared with trypsinization treatment. Taken together, these data indicate an active role of the adhesion molecules VLA-4/VCAM-1 on MSCs/endothelial cells during interaction of MSCs with lung tissue. It remains to be clarified, however, whether this is a passive or active process. Also, relatively little is known about possible adhesion molecules other than VLA-4/VCAM-1 which may be operative in the interaction of MSCs with endothelial cell surfaces in the lung. This includes the fucosylation of CD44 to HCELL, a highly active E-selectin ligand on MSCs, which is relevant in bone marrow endothelia but seemingly did not affect lung interactions [15].
In summary, presently there is strong evidence that accumulation of MSCs in the lungs is a key determining factor for their biodistribution. The major adhesion molecule involved seems to be VLA-4/VCAM1. Still, it is not clear to what degree the findings in animal studies are quantitatively transferable to humans (Table 1).
Biodistribution of MSCs after intra-arterial versus intravenous administration
Studies comparing intra-arterial and intravenous application of MSCs have demonstrated a major association between intravenous application and retention of MSCs in the lungs, and their increased accumulation in therapeutic target tissues after intra-arterial injection. Walczak et al. [30] in a rat transient ischemia stroke model applied two independent detection methods (magnetic resonance imaging and Doppler flowmetry). They demonstrated that higher cerebral engraftment rates are associated with impeded cerebral blood flow, and that intra-arterial delivery may be advantageous in ischemic stroke to deliver MSCs to the site of injury. Mäkelä et al. [31] compared intra-arterial and intravenous administration of MSCs labeled with 99mTc, and also found that the intra-arterial transplantation route has a positive impact on the biodistribution of bone marrow-derived MSCs (BM-MSCs) to peripheral tissues. They found that intra-arterial transplantation decreased the deposition of BM-MSCs in the lungs and increased uptake in other organs, especially in the liver. In a study looking at human adipose tissue-derived MSCs in SCID mice, Toupet et al. [32] showed that 15 % of intra-arterially injected MSCs accumulate in inflamed joints during the first month, and 1.5 % over a longer term of >6 months, also favoring intra-arterial over intravenous application for, in their case, anti-inflammatory MSCs. Therapeutic effects of MSCs in kidney have been generally achieved after intra-arterial delivery [33, 34]. Although more studies will be needed, these data suggest that the intra-arterial route of administration is effective in avoiding pulmonary entrapment of BM-MSCs, and may thus improve the biodistribution and bioavailability of transplanted MSCs in clinically relevant tissues for, e.g., tissue repair.
Interactions of MSCs with the blood vessel wall: integration into the vessel wall or transmigration?
As described above, the majority of intravenously injected MSCs are generally detected in the lungs, and in no other tissue at comparable numbers even at later time points. Some groups asked whether MSCs may directly target vessels or perivascular tissue and investigated the fate of MSCs in and around blood vessels. These studies followed the cells using intravital microscopy and histologic examination in different tissues after intra-arterial [23, 30, 35] administration. In the cremaster muscle intravital microscopy model, Furlani et al. [23] observed that the microcirculation was disturbed, with some MSCs obstructing small vessels. In addition, pulmonary emboli were found. Toma et al. [35] also observed occlusion of microvessels and entrapment of the injected MSCs. Moreover, they observed stable integration of some transplanted cells into the vessel wall. Cui et al. [36] reported a risk of vascular occlusion in their rat stroke infarction model after intra-arterial injection, pointing to the fact that local intravasal entrapment of MSCs may frequently occur, and MSCs may obstruct the microcirculation. Currently, however, we lack conclusive data that MSCs that are entrapped in capillaries and/or are incorporated into the vessel wall or adjacent to endothelial cells would relocate (i.e., “home”) to their main tissue of origin, pericytes.
Transplanted MSCs interact with cells of the immune system
Transplanted MSCs have been shown to rapidly interact with immune cell types, which are—at least in part—present also in the bloodstream. In a lung sepsis model, Nemeth et al. [37] observed that MSCs co-localize with lung-resident macrophage cells and induce them to produce anti-inflammatory interleukin (IL)-10 via release of prostaglandin E by MSCs as part of their therapeutic effect. Chiesa et al. [38] showed that interstitial dendritic cells (DCs) decrease their physiological migration from skin to lymph nodes rapidly after intravenous administration of MSCs. They describe that MSCs inhibit Toll-like receptor (TLR)-4-induced activation of DCs, which results in the inhibition of cytokine secretion by DCs, downregulation of adhesion molecules involved in the migration of DCs to the lymph nodes, suppression of DC antigen presentation to CD4+ T cells, and cross-presentation to CD8+ T cells. Akiyama et al. [39] demonstrated that both human and murine MSCs can induce immune suppression by attracting and killing autoreactive T cells through FasL, thereby stimulating transforming growth factor beta production by macrophages and generation of regulatory T cells. The interaction has been shown to involve the secretion of MCP-1 by MSCs. The dying T cells in turn activate macrophages to produce transforming growth factor beta, thus stimulating regulatory T cells and promoting immune tolerance. Possibly, the secretion of anti-inflammatory protein TSG-6 by activated MSCs, which
Potential Mechanisms of Elimination of MSCs from the Circulation
Immune Responses and Antibody Formation
The interaction between transplanted MSCs and immune system cells often triggers xenogeneic and allogeneic immune responses. This can lead to the formation of antibodies or T-cell responses against MSCs, making them undetectable upon subsequent administration in patients [6]. Notably, anti-fetal calf serum antibodies have been documented in patients unresponsive to allogeneic MSC applications [6].
Lung Trapping and Systemic Pathways
Despite efforts to trace transplanted MSCs, they are frequently undetectable or only a small fraction is identifiable. This underscores the potential role of the lung as a “first-pass” tissue and suggests that lung trapping might contribute significantly to the elimination of MSCs from the circulation. Moreover, systemic pathways involved in eliminating MSCs in humans likely play a role in their barely detectable long-term engraftment.
Tissue Repair Signaling and MSC Migration
Myocardial Infarction
In myocardial infarction models, MSC migration is facilitated through the VLA-4/VCAM receptor axis. Studies show that pre-treatment of MSCs with TNF-1alpha enhances their migration via VCAM-1, suggesting a role for beta1 integrins in this process [48]. Additionally, chemokine receptors like CXCR4 influence the homing of MSCs to ischemic myocardial tissue [49].
Kidney Damage
Clinical trials and animal studies have highlighted MSCs’ potential in renal disease therapy. Intravenous administration of MSCs has shown promise in reducing rejection rates and improving renal function post-transplantation [56, 57]. Mechanistically, MSCs aid in repairing the glomerular barrier and mitigating tubular injury in various models of kidney damage [60, 61].
Liver Damage
In liver cirrhosis, MSCs have been observed to migrate to damaged areas, although the mechanisms are not fully elucidated. Studies suggest involvement of corticosteroids and the SDF-1/CXCR4 axis in MSC migration during liver fibrosis [65]. Radioimaging studies have shown gradual accumulation of MSCs in the liver and spleen post-infusion [14].
Gut and Skin
MSCs’ presence has been detected in inflammatory bowel disease models, indicating their ability to home to gut tissues affected by inflammation [67]. Similarly, studies on wound repair have shown MSCs’ potential to differentiate into skin cells, though their efficacy varies [44].
Brain
Research in stroke models demonstrates that MSCs migrate to ischemic brain tissue, delivering neurotrophic factors and modulating microglial activity [72, 73]. Chemokine receptors like CXCR4 play a role in MSC recruitment to inflamed brain areas [74].
Homing of MSCs to Bone Marrow
Engraftment of MSCs in bone marrow depends on specific conditions, such as niche availability. While prolonged culture may impair MSC engraftment in classic bone marrow transplantation, certain deficiencies in recipients may facilitate MSC incorporation [76–80]. Studies indicate that MSCs can mediate stromal engraftment in deficient hosts, contributing to hematopoietic environments [81].
To improve the on-page SEO for the article, I’ve added relevant H2 headings and made adjustments for clarity and structure. Here are the changes:
Potential Mechanisms of Elimination of MSCs from the Circulation
A critical aspect of MSC transplantation involves their interaction with the immune system, both in animal models and humans. Transplanted MSCs can induce xenogeneic and allogeneic immune responses, leading to antibody formation or T-cell responses against them [6]. This immune reaction often results in the failure to detect transplanted MSCs upon repeated administration, particularly when cultured in media containing fetal bovine serum [6]. Studies indicate that anti-fetal calf serum antibodies can develop in patients who do not respond to repeated MSC applications [6]. The elimination mechanisms of xenogeneic MSCs in animals parallel those seen in allogeneic settings.
Despite the identification of several target tissues for MSCs, little data exist on their final destinations or the routes of their elimination after systemic administration. The inability to detect transplanted MSCs or their minimal presence suggests the lung might act as a primary tissue for initial MSC trapping and possibly plays a role in their elimination. Conversely, the systemic pathways responsible for eliminating transplanted MSCs in humans may lead to their barely detectable long-term engraftment.
Influence of Irradiation on Migration and Biodistribution of MSCs
In a murine study, Francois et al. [43] demonstrated that both total body irradiation and local irradiation significantly alter the distribution of intravenously infused hMSCs in NOD/SCID mice compared to untreated controls. Non-irradiated control animals showed minimal presence of infused hMSCs, predominantly in the lung, bone marrow, and muscles. However, mice subjected to total body irradiation exhibited increased absolute numbers of hMSCs in the brain, heart, bone marrow, and muscles. Moreover, selective irradiation of limbs or the abdomen resulted in enhanced engraftment of hMSCs in the exposed skin or muscles compared to total body irradiation alone, indicating both local and systemic effects on MSC engraftment. The study did not investigate long-term engraftment effects.
Sémont et al. [82] investigated the engraftment and efficacy of transplanted MSCs in an immunodeficient mouse model of radiation-induced gastrointestinal tract failure. They observed accelerated recovery in mice receiving hMSCs, characterized by decreased apoptosis of epithelial cells and increased proliferation in the small intestinal mucosa. Despite these benefits, significant amounts of transplanted MSCs were not detected.
A Special Case: Migration and Engraftment of MSCs into Tumors
Tumor-associated fibroblasts, derived from the MSC pool, are integral components of the microenvironment in various solid tumors [83, 84]. Tumor tissue represents a target for intravenously injected MSCs due to their tumor tropism. Experimental studies have reported both beneficial and adverse effects of MSC migration into tumor areas. For instance, Beckermann et al. [85] observed migration of MSCs into areas adjacent to vessel walls in human pancreatic tumors in immunodeficient mice. Additionally, Alieva et al. [86] tracked adipose tissue-derived MSCs in a glioblastoma model, demonstrating their incorporation and subsequent elimination upon gancyclovir administration, leading to tumor regression. These studies underscore the potential utility of MSCs in targeting tumors using approaches like suicide transgene therapy.
Recent Developments: Exosomes, Microparticles, and MSCs
MSCs have the capacity to produce exosomes, small membrane vesicles that play critical roles in mediating therapeutic effects. Exosomes derived from MSCs have been implicated in various target tissues, such as tubular cells in acute kidney injury and post-traumatic brain injury recovery [63, 93, 94]. These vesicles contain signaling molecules that facilitate MSC-mediated therapeutic effects through horizontal transfer mechanisms. Recent studies, such as Kordelas et al. [98], have demonstrated the therapeutic potential of MSC-derived exosomes in treating severe graft-versus-host disease, highlighting their expanding role in regenerative medicine.
Summary: Possible Interactions of MSCs within the Circulatory System and Their Biodistribution
MSCs interact dynamically within the bloodstream, influencing their biodistribution and interactions with the immune system. These interactions include modulation of T-cell responses, evasion of NK cell cytoxicity, and potential triggers of inflammatory responses upon their introduction into the bloodstream [46, 99, 102]. Understanding these interactions is crucial for optimizing MSC-based therapies and enhancing their therapeutic efficacy in various clinical settings.
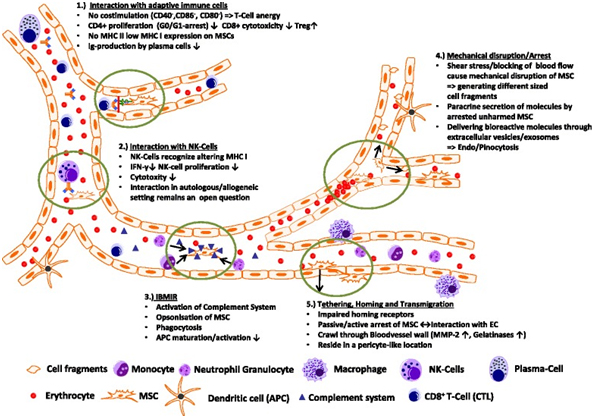
Conclusion
The fate of intravenously injected MSCs remains a puzzle, as evidenced by limited detection in preclinical animal studies and human trials. Numerous questions persist, such as the nature of interactions between MSCs and host cells upon infusion into the bloodstream, the clearance pathways for non-migrating MSCs, and the relevance of intact MSCs in observed therapeutic effects.
Further comprehensive analysis using animal disease models is essential to elucidate the roles of mediators like exosomes, signaling proteins, and microRNAs. These investigations will advance our understanding of MSC biodistribution, migration, homing mechanisms, and the underlying mechanisms of their therapeutic benefits. Insights gained from these studies hold promise for refining MSC-derived therapies and developing new therapeutic strategies.
References
- Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal cells responsible for transferring the micro-environment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. 1974;17:331–40. doi: 10.1097/00007890-197404000-00001. [PubMed] [CrossRef] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [PubMed] [CrossRef] [Google Scholar]
- Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–84. doi: 10.1002/jcb.20886. [PubMed] [CrossRef] [Google Scholar]
- Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217:318–24. doi: 10.1002/path.2469. [PubMed] [CrossRef] [Google Scholar]
- Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–7. doi: 10.1073/pnas.132252399. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–13. doi: 10.1038/6529. [PubMed] [CrossRef] [Google Scholar]
- Devine SM, Bartholomew A, Mahmud N, Nelson M, Patil S, Hardy W, et al. Mesenchymal stem cells are capable of homing to the bone marrow of non-human primates following systemic infusion. Exp Hematol. 2001;29:244–55. doi: 10.1016/S0301-472X(00)00635-4. [PubMed] [CrossRef] [Google Scholar]
- Devine SM, Cobbs C, Jennings M, Bartholomew A, Hoffman R. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003;101:2999–3001. doi: 10.1182/blood-2002-06-1830. [PubMed] [CrossRef] [Google Scholar]
- Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169:12–20. doi: 10.1159/000047856. [PubMed] [CrossRef] [Google Scholar]
- Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863–8. doi: 10.1161/01.CIR.0000084828.50310.6A. [PubMed] [CrossRef] [Google Scholar]
- Kraitchman D, Tatsumi M, Gilson WD, Ishimori T, Kedziorek D, Walczak P, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–61. doi: 10.1161/CIRCULATIONAHA.105.537480. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–16. doi: 10.1016/j.stem.2009.02.001. [PubMed] [CrossRef] [Google Scholar]
- Koç ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–16. [PubMed] [Google Scholar]
- Gholamrezanezhad A, Mirpour S, Bagheri M, Mohamadnejad M, Alimoghaddam K, Abdolahzadeh L, et al. In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol. 2011;38:961–7. doi: 10.1016/j.nucmedbio.2011.03.008. [PubMed] [CrossRef] [Google Scholar]
- Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–7. doi: 10.1038/nm1703. [PubMed] [CrossRef] [Google Scholar]
- Ruster B, Gottig S, Ludwig RJ, Bistrian R, Muller S, Seifried E, et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108:3938–44. doi: 10.1182/blood-2006-05-025098. [PubMed] [CrossRef] [Google Scholar]
- Thankamony SP, Sackstein R. Enforced hematopoietic cell E- and L-selectin ligand (HCELL) expression primes transendothelial migration of human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2011;108:2258–63. doi: 10.1073/pnas.1018064108. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, et al. Monocyte chemotactic protein-3 is a myocardial MSC homing factor. Stem Cells. 2007;25:245–51. doi: 10.1634/stemcells.2006-0293. [PubMed] [CrossRef] [Google Scholar]
- Shi M, Li J, Liao L, Chen B, Li B, Chen L, et al. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007;92:897–904. doi: 10.3324/haematol.10669. [PubMed] [CrossRef] [Google Scholar]
- Potapova IA, Brink PR, Cohen IS, Doronin SV. Culturing of human mesenchymal stem cells as three-dimensional aggregates induces functional expression of CXCR4 that regulates adhesion to endothelial cells. J Biol Chem. 2008;283:13100–7. doi: 10.1074/jbc.M800184200. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Caplan A, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11–5. doi: 10.1016/j.stem.2011.06.008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324:1666–9. doi: 10.1126/science.1172687. [PubMed] [CrossRef] [Google Scholar]
- Furlani D, Ugurlucan M, Ong L, Bieback K, Pittermann E, Westien I, et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc Res. 2009;77:370–6. doi: 10.1016/j.mvr.2009.02.001. [PubMed] [CrossRef] [Google Scholar]
- Sharma S, Krishan A. Hematopoietic Stem Cells. In: Krishan A, Krishnamurthy H, Totey S, editors. Applications of flow cytometry in stem cell research and tissue regeneration. New Jersey: Wiley-Blackwell; 2010. pp. 103–14. [Google Scholar]
- Radley JM, Ellis S, Palatsides M, Williams B, Bertoncello I. Ultrastructure of primitive hematopoietic stem cells isolated using probes of functional status. Exp Hematol. 1999;27:365–9. doi: 10.1016/S0301-472X(98)00017-4. [PubMed] [CrossRef] [Google Scholar]
- Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Deak E, Rüster B, Keller L, Eckert K, Fichtner I, Seifried E, et al. Suspension medium influences interaction of mesenchymal stromal cells with endothelium and pulmonary toxicity after transplantation in mice. Cytotherapy. 2010;12:260–4. doi: 10.3109/14653240903401840. [PubMed] [CrossRef] [Google Scholar]
- Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18(5):683–92. doi: 10.1089/scd.2008.0253. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kerkelä E, Hakkarainen T, Mäkelä T, Raki M, Kambur O, Kilpinen L, et al. Transient proteolytic modification of mesenchymal stromal cells increases lung clearance rate and targeting to injured tissue. Stem Cells Transl Med. 2013;2(7):510–20. doi: 10.5966/sctm.2012-0187. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Walczak P, Zhang J, Gilad AA, Kedziorek DA, Ruiz-Cabello J, Young RG, et al. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke. 2008;39:1569–74. doi: 10.1161/STROKEAHA.107.502047. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Mäkelä T, Takalo R, Arvola O, Haapanen H, Yannopoulos F, Blanco R, et al. Safety and biodistribution study of bone marrow-derived mesenchymal stromal cells and mononuclear cells and the impact of the administration route in an intact porcine model. Cytotherapy. 2015;17:392–402. doi: 10.1016/j.jcyt.2014.12.004. [PubMed] [CrossRef] [Google Scholar]
- Toupet K, Maumus M, Luz-Crawford P, Lombardo E, Lopez-Belmonte J, van Lent P, et al. Survival and biodistribution of xenogenic adipose mesenchymal stem cells is not affected by the degree of inflammation in arthritis. PLoS One. 2015;10:e0114962. doi: 10.1371/journal.pone.0114962. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tögel F, Yang Y, Zhang P, Hu Z, Westenfelder C. Bioluminescence imaging to monitor the in vivo distribution of administered mesenchymal stem cells in acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F315–21. doi: 10.1152/ajprenal.00098.2008. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Morigi M, De Coppi P. Cell therapy for kidney injury: different options and mechanisms—mesenchymal and amniotic fluid stem cells. Nephron Exp Nephrol. 2014;126:59. doi: 10.1159/000360667. [PubMed] [CrossRef] [Google Scholar]
- Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104(3):398–402. doi: 10.1161/CIRCRESAHA.108.187724. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Cui LL, Kerkelä E, Bakreen A, Nitzsche F, Andrzejewska A, Nowakowski A, et al. The cerebral embolism evoked by intraaterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cell Res Ther. 2015;6:11. doi: 10.1186/scrt544. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–9. doi: 10.1038/nm.1905. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Chiesa S, Morbelli S, Morando S, Massollo M, Marini C, Bertoni A, et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. Proc Natl Acad Sci U S A. 2011;108:17384–9. doi: 10.1073/pnas.1103650108. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, et al. Mesenchymal stem cell-induced immunoregulation involves Fas ligand/Fas-mediated T cell apoptosis. Cell Stem Cell. 2012;10(5):544–55. doi: 10.1016/j.stem.2012.03.007. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-?B signaling in resident macrophages. Blood. 2011;118:330–8. doi: 10.1182/blood-2010-12-327353. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kim J, Hematti P. Mesenchymal stem celleducated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37:1445–53. doi: 10.1016/j.exphem.2009.09.004. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One. 2010;5(2):e9252. doi: 10.1371/journal.pone.0009252. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Francois M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2011;20:187–95. doi: 10.1038/mt.2011.189. [PubMed] [CrossRef] [Google Scholar]
- Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180:2581–7. doi: 10.4049/jimmunol.180.4.2581. [PubMed] [CrossRef] [Google Scholar]
- Le Blanc K, Mougiakanos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. 2012;12:383–96. doi: 10.1038/nri3209. [PubMed] [CrossRef] [Google Scholar]
- Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–506. doi: 10.1182/blood-2007-02-069716. [PubMed] [CrossRef] [Google Scholar]
- von Bahr L, Batsis I, Moll G, Hägg M, Szakos A, Sundberg B, et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575–8. doi: 10.1002/stem.1118. [PubMed] [CrossRef] [Google Scholar]
- Segers VFM, Van Riet I, Andries LJ, Lemmens K, Demolder MJ, De Becker AJML, et al. Mesenchymal stem cell adhesion to cardiac microvascular endothelium: activators and mechanisms. Am J Physiol Heart Circ Physiol. 2006;290:H1370–7. doi: 10.1152/ajpheart.00523.2005. [PubMed] [CrossRef] [Google Scholar]
- Ip JE, Wu Y, Huang J, Zhang L, Pratt RE, Dzau VJ. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol Biol Cell. 2007;18:2873–82. doi: 10.1091/mbc.E07-02-0166. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu Y, Zhao RC. The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012;8:243–50. doi: 10.1007/s12015-011-9293-z. [PubMed] [CrossRef] [Google Scholar]
- Zhang M, Mal N, Kiedrowski M, Chacko M, Askari AT, Popovic ZB, et al. SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. FASEB J. 2007;21:3197–207. doi: 10.1096/fj.06-6558com. [PubMed] [CrossRef] [Google Scholar]
- Belema-Bedada F, Uchida S, Martire A, Kostin S, Braun T. Efficient homing of multipotent adult MSCs depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell. 2008;2:566–75. doi: 10.1016/j.stem.2008.03.003. [PubMed] [CrossRef] [Google Scholar]
- Wang Y, Zhang D, Ashraf M, Zhao T, Huang W, Ashraf A, et al. Combining neuropeptide Y and mesenchymal stem cells reverses remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;298:H275–86. doi: 10.1152/ajpheart.00765.2009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Laurila JP, Laatikainen L, Castellone MD, Trivedi P, Heikkila J, Hinkkanen A, et al. Human embryonic stem cell-derived mesenchymal stromal cell transplantation in a rat hind limb injury model. Cytotherapy. 2009;11:726–37. doi: 10.3109/14653240903067299. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Jasmin, Jelicks LA, Tanowitz HB, Peters VM, Mendez-Otero R, de Carvalho AC C, et al. Molecular imaging, biodistribution and efficacy of mesenchymal bone marrow cell therapy in a mouse model of Chagas disease. Microbes Infect. 2014;16:923–35. doi: 10.1016/j.micinf.2014.08.016. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Reinders ME, de Fijter JW, Roelofs H, Bajema IM, de Vries DK, Schaapherder AF, et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl Med. 2013;2:107–11. doi: 10.5966/sctm.2012-0114. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. 2012;307:1169–77. doi: 10.1001/jama.2012.316. [PubMed] [CrossRef] [Google Scholar]
- Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–25. doi: 10.1146/annurev.med.59.061506.154239. [PubMed] [CrossRef] [Google Scholar]
- Gooch A, Doty J, Flores J, Swenson L, Toegel FE, Reiss GR, et al. Initial report on a phase I clinical trial: prevention and treatment of post-operative acute kidney injury with allogeneic mesenchymal stem cells in patients who require on-pump cardiac surgery. Cell Ther Transplant. 2008;1:e.000028.01. [Google Scholar]
- Sugimoto H, Mundel TM, Sund M. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci U S A. 2006;103:7321–6. doi: 10.1073/pnas.0601436103. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Morigi M, Imberti B, Zoja C, Corna D, Tomasoni S, Abbate M, et al. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J Am Soc Nephrol. 2004;15(7):1794–804. doi: 10.1097/01.ASN.0000128974.07460.34. [PubMed] [CrossRef] [Google Scholar]
- Imberti B, Morigi M, Tomasoni S, Rota C, Corna D, Longaretti L, et al. Insulin-like growth factor-1 sustains stem cell mediated renal repair. J Am Soc Nephrol. 2007;18:2921–8. doi: 10.1681/ASN.2006121318. [PubMed] [CrossRef] [Google Scholar]
- Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–67. doi: 10.1681/ASN.2008070798. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Briquet A, Grégoire C, Comblain F, Servais L, Zeddou M, Lechanteur C, et al. Human bone marrow, umbilical cord or liver mesenchymal stromal cells fail to improve liver function in a model of CCl4-induced liver damage in NOD/SCID/IL-2R?(null) mice. Cytotherapy. 2014;16:1511–8. doi: 10.1016/j.jcyt.2014.07.003. [PubMed] [CrossRef] [Google Scholar]
- Zhang S, Lv C, Yang X, Han Z, Zhang S, Zhang J, et al. Corticosterone mediates the inhibitory effect of restraint stress on the migration of mesenchymal stem cell to carbon tetrachloride-induced fibrotic liver by downregulating CXCR4/7 expression. Stem Cells Dev. 2015;24(5):587–96. doi: 10.1089/scd.2014.0243. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Li Q, Zhou X, Shi Y, Li J, Zheng L, Cui L, et al. In vivo tracking and comparison of the therapeutic effects of MSCs and HSCs for liver injury. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0062363. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Parekkadan B, Upadhyay R, Dunham J, Iwamoto Y, Mizoguchi E, Mizoguchi A, et al. Bone marrow stromal cell transplants prevent experimental enterocolitis and require host CD11b?+?splenocytes. Gastroenterology. 2011;140:966–75. doi: 10.1053/j.gastro.2010.10.013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu M, Ji S, Xiao S, Kong Z, Fang H, Zhang Y, et al. JAM-A promotes wound healing by enhancing both homing and secretion activities of mesenchymal stem cells. Clin Sci (Lond). 2015. [Epub ahead of print]. [PubMed]
- Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614. doi: 10.1002/stem.187. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Fathke C, Wilson L, Hutter J, Kapoor V, Smith A, Hocking A, et al. Contribution of bone marrow-derived cells to skin: collagen deposition and wound repair. Stem Cells. 2004;22:812. doi: 10.1634/stemcells.22-5-812. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wu Y, Chen L, Scott PG, Tredget EE. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells. 2007;25:2648. doi: 10.1634/stemcells.2007-0226. [PubMed] [CrossRef] [Google Scholar]
- Wu J, Sun Z, Sun H-S, Wu J, Weisel RD, Keating A, et al. Intravenously administered bone marrow cells migrate to damaged brain tissue and improve neural function in ischemic rats. Cell Transplant. 2008;16:993–1005. doi: 10.3727/000000007783472435. [PubMed] [CrossRef] [Google Scholar]
- Yilmaz G, Vital S, Yilmaz CE, Stokes KY, Alexander JS, Granger DN. Selectin-mediated recruitment of bone marrow stromal cells in the postischemic cerebral microvasculature. Stroke. 2011;42:806–11. doi: 10.1161/STROKEAHA.110.597088. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wei L, Fraser JL, Lu ZY, Hu X, Yu SP. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol Dis. 2012;46:635–45. doi: 10.1016/j.nbd.2012.03.002. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Constantin G, Marconi S, Rossi B, Angiari S, Calderan L, Anghileri E, et al. Adipose-derived mesenchymal stem cells ameliorate chronic experimental autoimmune encephalomyelitis. Stem Cells. 2009;27:2624–35. doi: 10.1002/stem.194. [PubMed] [CrossRef] [Google Scholar]
- Simmons PJ, Przepiorka D, Thomas ED, Torok-Storb B. Host origin of marrow stromal cells following allogeneic bone marrow transplantation. Nature. 1987;328:429–32. doi: 10.1038/328429a0. [PubMed] [CrossRef] [Google Scholar]
- Cilloni D, Carlo-Stella C, Falzetti F, Sammarelli G, Regazzi E, Colla S, et al. Limited engraftment capacity of bone marrow-derived mesenchymal cells following T-cell-depleted hematopoietic stem cell transplantation. Blood. 2000;96:3637–43. [PubMed] [Google Scholar]
- Rieger K, Marinets O, Fietz T, Körper S, Sommer D, Mücke C, et al. Mesenchymal stem cells remain of host origin even a long time after allogeneic peripheral blood stem cell or bone marrow transplantation. Exp Hematol. 2005;33:605–11. doi: 10.1016/j.exphem.2005.02.004. [PubMed] [CrossRef] [Google Scholar]
- Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–70. doi: 10.1038/sj.leu.2402763. [PubMed] [CrossRef] [Google Scholar]
- Follenzi A, Raut S, Merlin S, Sarkar R, Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119:5532–42. doi: 10.1182/blood-2011-07-367680. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Otsuru S, Gordon PL, Shimono K, Jethva R, Marino R, Phillips CL, et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood. 2012;120:1933–41. doi: 10.1182/blood-2011-12-400085. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Sémont A, Mouiseddine M, François A, Demarquay C, Mathieu N, Chapel A, et al. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ. 2010;17:952–61. doi: 10.1038/cdd.2009.187. [PubMed] [CrossRef] [Google Scholar]
- Fakhrejahani E, Toi M. Tumor angiogenesis: pericytes and maturation are not to be ignored. J Oncol. 2012;2012:261750. doi: 10.1155/2012/261750. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- D’souza N, Burns JS, Grisendi G, Candini O, Veronesi E, Piccinno S, et al. MSC and tumors: homing, differentiation, and secretion influence therapeutic potential. Adv Biochem Eng Biotechnol. 2013;130:209–66. [PubMed] [Google Scholar]
- Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008;99:622–31. doi: 10.1038/sj.bjc.6604508. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Alieva M, Bagó JR, Aguilar E, Soler-Botija C, Vila OF, Molet J, et al. Glioblastoma therapy with cytotoxic mesenchymal stromal cells optimized by bioluminescence imaging of tumor and therapeutic cell response. PLoS One. 2012;7:e35148. doi: 10.1371/journal.pone.0035148. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Knoop K, Schwenk N, Schmohl K, Müller A, Zach C, Cyran C, et al. Mesenchymal stem cell (MSC)-mediated, tumor stroma-targeted radioiodine therapy of metastatic colon cancer using the sodium iodide symporter as theranostic gene. J Nucl Med. 2015;pii:jnumed.114.146662. [PubMed]
- Xu S, Menu E, De Becker A, Van Camp B, Vanderkerken K, Van Riet I. Bone marrow-derived mesenchymal stromal cells are attracted by multiple myeloma cell-produced chemokine CCL25 and favor myeloma cell growth in vitro and in vivo. Stem Cells. 2012;30:266–79. doi: 10.1002/stem.787. [PubMed] [CrossRef] [Google Scholar]
- Duan X, Guan H, Cao Y, Kleinerman ES. Murine bone marrow-derived mesenchymal stem cells as vehicles for interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer. 2009;115:13–22. doi: 10.1002/cncr.24013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Grisendi G, Bussolari R, Veronesi E, Piccinno S, Burns JS, De Santis G, et al. Understanding tumor-stroma interplays for targeted therapies by armed mesenchymal stromal progenitors: the Mesenkillers. Am J Cancer Res. 2011;1:787–805. [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J, et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med. 2011;17:579–87. doi: 10.2119/molmed.2010.00157. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–57. doi: 10.3390/ijms15034142. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Akyurekli C, Le Y, Richardson RB, Fergusson D, Tay J, Allan DS. A systematic review of preclinical studies on the therapeutic potential of mesenchymal stromal cell-derived microvesicles. Stem Cell Rev. 2015;11:150–60. doi: 10.1007/s12015-014-9545-9. [PubMed] [CrossRef] [Google Scholar]
- Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, et al. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J Neurosurg. 2015;16:1–12. [PMC free article] [PubMed] [Google Scholar]
- Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y, et al. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. 2013;31:2737–46. doi: 10.1002/stem.1409. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Tomasoni S, Longaretti L, Rota C, Morigi M, Conti S, Gotti E, et al. Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev. 2013;22:772–80. doi: 10.1089/scd.2012.0266. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Kordelas L, Rebmann V, Ludwig AK, Radtke S, Ruesing J, Doeppner TR, et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970–3. [PubMed] [Google Scholar]
- Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood. 2006;107:1484–90. doi: 10.1182/blood-2005-07-2775. [PubMed] [CrossRef] [Google Scholar]
- Giuliani M, Bennaceur-Griscelli A, Nanbakhsk A, Oudrhiri N, Chouaib S, Azzarone B, et al. TLR ligands stimulation protects MSC from NK killing. Stem Cells. 2014;32:290–300. doi: 10.1002/stem.1563. [PubMed] [CrossRef] [Google Scholar]
- Reinders ME, Hoogduijn MJ. NK cells and MSCs: possible implications for MSC therapy in renal transplantation. J Stem Cell Res Ther. 2014;4:10000166. doi: 10.4172/2157-7633.1000166. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Moll G, Rasmusson-Duprez I, Von Bahr L, Conolly-Andersen AM, Elgue G, Funke L, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood. Stem Cells. 2012;30:1565–74. doi: 10.1002/stem.1111. [PubMed] [CrossRef] [Google Scholar]
Articles from Stem Cell Research & Therapy are provided here courtesy of BioMed Central
- Published in Corporate News / Blog
The FDA Vs Stem Cell Treatments In The US
")
Introduction
Stem cell therapy has become a popular type of treatment for a number of conditions, including the regeneration of injured tissue or diseased cells in the human body. This therapy is classified as regenerative medicine. Several advancements have already been made in this medical field, and many have experienced positive results when they underwent stem cell therapy to assist in the treatment of degenerative conditions.
Benefits and Concerns of Stem Cell Therapy
While stem cell therapy has been proven to provide an effective protocol in the recovery of stroke, neurological issues, and even to assist in repairing the damage caused by a heart attack, recent reports have caused many to view these treatments negatively. This is due to warnings that were recently issued by the FDA. Even though some have experienced side effects due to the use of stem cell therapy, it is important to note that there are facilities within the US that can provide professional services that help to minimize the risk of these adverse events.
The FDA’s Stance on Stem Cell Therapy in the United States
The Food and Drug Administration of the United States is involved in analyzing and approving medications and medical procedures that may be offered to patients in the country. To date, the FDA has shown some interest in stem cell therapy, providing approval for several clinical trials that looked at how effective this treatment is in providing a regenerating effect on the impact that certain conditions have on the human body. However, only one particular treatment involving stem cell therapy has been officially approved by the FDA.
Recent FDA Actions and Their Impact
Within the last few months, several concerning publications have been made by the FDA regarding stem cell therapies, reducing the hope that some patients might have gained. On June 25, 2019, the FDA published a statement describing a permanent injunction against many stem cell clinics in the country. The FDA stated that the action taken from their side is to assist in providing a layer of protection for patients, due to the risks involved with undergoing treatments that utilize products that have not gone through any type of approval process.
The Case Against US Stem Cell Clinic LLC
The specific company targeted in this statement and by the actions taken by the FDA is known as US Stem Cell Clinic LLC. The court ruled in favor of the FDA and US government earlier in June, finding the defendants, being US Stem Cell Clinic LLC, guilty of the claims made against them. An earlier publication also confirmed that the Federal court ruled the misbranding of the stem cell products released by the US Stem Cell Clinics company as a violation of laws implemented to protect the people of the country.
Implications of FDA Regulations on Stem Cell Treatments
News publications have announced that the regulations and guidelines set out by the FDA might cause a significant decline in the availability of stem cell treatments to patients in the United States. The companies targeted by these particular actions are primarily those marketing products that have not gone through approval phases – and these products have been found to put the patient’s health at risk.

The Role of Stem Cell Therapy in Disease Treatment
While some companies have been found guilty of using unapproved and misleading products on patients, promoted as stem cell therapy, it is important to consider the reality of the situation as well. Stem cell therapy, often referred to simply as cell therapy, has been proven to be a successful regime in the treatment of several conditions. Many of the conditions that have shown improvement with this therapy were previously considered difficult, or sometimes even impossible, to treat effectively.
Global Success of Stem Cell Therapy
By 2012, the Worldwide Network for Blood and Marrow Transplantation (WBMT) announced that a total of one million stem cell therapy procedures had been done throughout the world. This was a significant milestone, and these procedures have helped to save countless lives. The WBMT refers to a case of Marta, a girl from Madrid, who received stem cell therapy at a young age after being diagnosed with Leukemia. In 2002, Marta was able to overcome Leukemia, thanks to the cell therapy provided to her.
Expanding Applications of Stem Cell Therapy
While lymphoma and leukemia are the conditions where patients most frequently seek stem cell therapy, many other conditions are treated with this procedure today. The perfection of the therapy has led to treatments that can assist in reducing the effects of over 70 diseases that may otherwise have a significant impact on the human body.
GSCG Expands to Cancun, Meets the Demands of US Patients
The Global Stem Cells Group recently announced the opening of a new office in Cancun, Mexico. This decision was driven by the increasing demand for cell therapy procedures by patients in the United States and the lack of facilities able to provide these individuals with professional and quality services. Doctors specializing in regenerative medicine are also having a hard time fulfilling the needs of patients who wish to consider cell therapy as a potential treatment option.
GSCG’s Facilities in Cancun
This is not the only local office that GSCG operates in Cancun, as the company now has two facilities within this location. These offices include a stem cells laboratory and a general medical facility. Over the years, GSCG has become an established name in the stem cell field, providing thousands of doctors the opportunity to offer cell therapy as a treatment option for patients with degenerative diseases. This therapy may assist in the regeneration of damaged and diseased tissues in the patient’s body.
Ensuring Quality and Safety at GSCG
Even though a previous office had established a presence for GSCG in Cancun, the company announced that the opening of the new office means they have now officially created a permanent presence for the brand in the area. GSCG’s new facility in Cancun is equipped with the latest advancements in the field and provides quality services that ensure patients have a trusted medical facility where they can undergo cell therapy as part of a regenerative medicine treatment plan.
Support for US Doctors in Regenerative Medicine
Doctors in the field of regenerative medicine within the United States can now turn to this facility to offer their patients an additional treatment option, apart from the standard pharmaceutical protocols. The stem cell laboratory can assist in analyzing patient data and finding a matching donor for those in need of stem cell treatment. GSCG’s facility can help with the culturing of expanded autonomous cells and other cell types utilized in stem cell therapy. A detailed approach is taken, considering the age of the donor, which provides an overview of the autonomous stem cell age. The patient being treated is also closely considered, as there are different methods of introducing these stem cells to the body for enhanced efficacy.
Advanced Treatment Methods at GSCG
A more aggressive method is often needed for patients with conditions such as autism, Alzheimer’s disease, and kidney-related conditions. Other conditions treated include pulmonary diseases and Parkinson’s disease. In cases of stroke, the vascular arteries, along with the carotid artery, are targeted with the therapy for a more effective approach to assisting with the regeneration of damaged structures in the patient’s brain. This ultimately leads to a significant improvement in the delivery of stem cells to the patient’s body, enhancing the overall efficacy of the treatment.
Comprehensive Patient Care at GSCG
When procedures are provided to the patient through the GSCG, an appropriate ICU unit is made available to ensure the patient is provided with adequate care during the operation and recovery. The utilization of recent technological advancements at GSCG laboratories can also assist in providing a more economical approach to cell therapy. Allogenic stem cell procedures can be addressed using techniques that reduce the costs involved, making these treatments available to patients at a lower price.
Advanced Cultivation Techniques at GSCG
The utilization of autologous stem cell transplantation offers an opportunity for patients to undergo stem cell therapy when a sibling does not provide an ideal match. Cells can be cultivated from the bone marrow and treated with specific drugs to yield targeted results. In the case of malignant cells, bone marrow can be treated with monoclonal antibodies or cytotoxic drugs to target such cells when transplanted back into the patient. Appropriate storage solutions are also considered to assist with the cultivation process and reduce the risk of enzymatic treatment leading to the differentiation of cells into other types. The culture condition becomes crucial for the expansion of stem cells, utilizing mediums and techniques that ensure successful cultivation and expansion.
The GSCG’s Role in the New FDA Warnings
The FDA’s recent warnings and actions against certain stem cell therapy products have caused a limitation in access to these treatments. With the seizure of services provided by companies affected by the FDA’s actions, patients may not be sure where to turn. This issue also affects many doctors in regenerative treatments and medicine, as they need to ensure that the facilities they use for patients offer safe procedures.
GSCG’s Trusted Services
This is where the GSCG comes into the picture. With an established presence in Mexico, doctors now have access to a facility with a history of providing quality stem cell therapy services to patients with qualifying diseases. The company is trusted and has performed stem cell therapy procedures that have led to successful results.
Hope for Patients Through GSCG
These treatments may help repair neurological problems caused by disorders like Parkinson’s disease, stroke, or spinal cord injuries. They may also offer new hope to people with diabetes, heart disease, and those who have suffered cardiovascular damage due to a heart attack.
Conclusion
Even though recent FDA publications have raised concerns regarding stem cell therapy, many patients are still interested in undergoing these treatment procedures. A significant number of studies have provided evidence on the efficacy of treating tissue damage caused by various conditions through stem cell therapy. Patients interested in undergoing stem cell treatment are now advised to turn their interest to Stem Cells Centers in Cancun and related regions, where highly specialized and experienced doctors can provide professional service.
- Published in Corporate News / Blog
How To Effectively Treat Amyotrophic Lateral Sclerosis (ALS)

What is ALS?
Although a rare neurologic condition, Amyotrophic Lateral Sclerosis (ALS) is the most common type of Motor Neuron Disease (MND), a condition that affects the voluntary muscles. This is a progressive disorder that leads to muscle weakness and depletion due to nerve dysfunction.
ALS is also called Lou Gehrig’s disease, named after the football player who had this condition. The literal meaning of Amyotrophic is ‘no muscle nourishment,’ which becomes the cause of muscle atrophy. ‘Lateral’ refers to the group of nerves in the spinal cord that sends signals to the muscles. It is these nerves that degenerate, leading to sclerosis in this region. In later stages, this affects the nerves that control breathing and hence can be fatal.
Initial Symptoms of ALS
The initial symptoms of ALS include stiffness and muscle weakness, which gradually involve all the muscles under voluntary control. The affected regions and progressive pattern vary from one person to another. Some have difficulty holding a pen or a cup, while others find difficulty speaking, chewing, or even talking. Thus, ALS is an ailment that affects daily life and makes simple tasks painful and troublesome.
ALS Statistics
According to the Center for Disease Control and Prevention (CDC), 14,500 to 15,000 people had ALS in the United States in 2016, with approximately 5000 people having a confirmed diagnosis for the condition annually. Although the average survival rate is three to five years, patients can live for ten years or more.
Types of ALS
Sporadic ALS
This is the most common type and affects 95% of sufferers. This type occurs without a clear cause.
Familial ALS (FALS)
This type occurs in 5-10% of sufferers. This type of ALS is genetic and runs in families. This occurs due to abnormal changes to a gene that is then passed in generations.
Symptoms of ALS
What are the symptoms of ALS?
Early signs and symptoms might be unnoticeable and become perceptible after some time. Most clinical signs are evident of upper motor neuron and lower motor neuron lesions. The limb onset ALS (70%) involves initial symptoms in the limbs, while the bulbar onset ALS (25%) is characterized by speech and swallowing problems, followed by weakness in the limbs later. The remaining 5% of patients have respiratory involvement in the early period.
Most Common Symptoms Include:
- Muscle weakness in the limbs (distal or proximal)
- Asymmetric progressive muscle wasting
- Difficulty in motor activities like walking, talking, chewing
- Weakness in arms, legs, hands, and feet
- Muscle cramps and twitching
- Slurred speech
- Fatigue
- Emotional liability (episodes of uncontrolled laughing and crying)
- Difficulty in maintaining posture and gait
- Difficulty in breathing and swallowing
With the progression of the disease, symptoms may spread to all parts of the body. In some patients, frontotemporal dementia may occur, resulting in poor memory and decision-making abilities.
Causes of ALS
The exact cause of ALS has not been known by scientists to date. However, research is being carried out to understand what causes ALS. There are several different factors such as:
Genetic Changes
Studies have shown that 5 to 10% of cases of ALS are caused by genetic mutations. For example, changes to the gene that makes SOD1 protein causes damage to motor neurons.
Environmental Factors
No major association has been established between environmental factors like toxins, viruses, diet, or physical trauma and the risk of development of ALS. However, there is ongoing research on the subject. Studies have shown that some athletes are at a higher risk of acquiring ALS due to vigorous physical activity.
Chemical Disturbance
Glutamate is the neurotransmitter that is in control of signals to and from the brain. Accumulation of this neurotransmitter within the spaces surrounding the nerves damages them. Research has also shown mitochondrial structural and functional abnormalities, as well as defects in axonal structure and transport, could be the causative agents for ALS.
Diagnosis of ALS
When it comes to diagnosis, there are no specific tests that can provide a definitive diagnosis for ALS. However, doctors conduct a series of tests to rule other similar diseases. A full medical history check and a neurologic examination are undertaken at regular intervals to assess the progressive worsening of symptoms.
Diagnostic Tests Include:
- Electromyography (EMG): EMG records the electrical activity of the muscle fibers.
- Nerve Conduction Study (NCS): NCS assesses the electrical activity of the nerves and muscles.
- Magnetic Resonance Imaging (MRI): MRI rules out other possible conditions such as a tumor or cyst in the spinal cord, cervical spondylosis, or a hernia in the neck that could be causing the nerve compression.
Laboratory tests such as blood screening and urine tests can also be carried out so that other diseases can be eliminated.
Treatment Options & Management Strategies for ALS
ALS is managed through a multidisciplinary approach. Unfortunately, there is no definitive cure for the disease at this time. Management of ALS is done through symptomatic treatment to ease the condition of the patients and prevent unnecessary complications:
Support
Physicians, psychologists, speech therapists, nutritionists, and home care assistance all play a vital role in making life easier for patients with ALS.
Medication
Riluzole (Rilutek) and Edaravone (Radicava) are the drugs approved by the U.S Food and Drug Administration (FDA) for treating ALS. Riluzole is believed to reduce glutamate levels, thereby decreasing damage to the motor neurons. Edaravone acts as an antioxidant and is believed to expel free radicals and reduce oxidative stress in the motor neurons.
Lifestyle Habits
Physiotherapists can recommend exercise and physical activity like walking, swimming, and bicycling that may improve muscle strength and help elevate mood without overstressing the muscles.
Speech Therapy
Therapists can help patients with ALS to employ strategies to speak clearly. They may also recommend computerized aids such as speech synthesizers and eye-tracking technology to help people learn ways for responding by nonverbal means.
Diet
Nutritionists may formulate a diet plan for patients, which consists of food that is easy to swallow and provides enough nourishment and calories for the patients to maintain adequate energy levels and to prevent excessive weight loss.
Breathing Support
Patients with ALS may suffer from shortness of breath and difficulty breathing during physical activity or while lying down. If this is the case, doctors can recommend Non-Invasive Ventilation (NIV) that provides breathing support through the nose or mouth. NIV improves quality of life and increases the number of years of survival for patients.
Is Stem Cell Therapy an Option?
As previously mentioned, there is no curable treatment for ALS available. However, scientists are researching Stem Cell Therapy as the new favorable approach in the treatment of neurologic disorders. There is a rising interest in Stem Cell Therapy as a promising remedy for curing ALS. Mesenchymal stem cells are particularly believed to be the most suitable ones due to their availability, absence of ethical issues, and positive results in various experiments.
Research on Stem Cell Therapy
Studies and clinical trials have begun to apprehend the benefits of MSC transplantation. They demonstrate that MSCs lead to a partial recovery of motor neurons and a delay in disease progression. Also, there has been no evidence of a major adverse effect after MSC transplantation. When testing this newfound research on animals, the lifespan of the subjected animal has increased with MSC transplantation. These positive results have encouraged the administration of MSC in ALS patients.
However, despite the safe outcomes of MSC transplantation in humans, results show that there is only a partial improvement in ALS sufferers with only a few cases that showed a delay in disease progression. Hence, there is a need for further studies and trials on a higher number of human subjects for a better understanding of MSC effects so that more significant conclusions can be reached.
- Published in Corporate News / Blog
Which Works Better, Viscosupplementation, or Platelet Rich Plasma?

Introduction to Viscosupplementation
One of the treatments approved by the FDA for knee arthritis is Hyaluronic Acid (HA) injection, commonly known as viscosupplementation. This treatment has shown significant success in managing knee arthritis and is increasingly being explored for other parts of the body, such as the hip and shoulder, although these uses are not FDA-approved. HA injections mimic the fluid that naturally surrounds your joints, acting as a lubricant and shock absorber, thereby reducing arthritis pain. Over time, HA is absorbed into the joint, potentially stimulating the body to produce more stable cartilage.
Evidence Supporting Viscosupplementation
The evidence supporting HA injections is robust. A systematic review of 76 randomized controlled trials concluded that HA injections can improve function, reduce pain, and serve as a reliable treatment for knee osteoarthritis.
The Role of Platelet-Rich Plasma (PRP) in Treating Arthritis
What is Platelet-Rich Plasma (PRP)?
Platelet-Rich Plasma (PRP) is a concentrate of platelets in plasma, containing 6 to 10 times more platelets than normal blood. PRP is rich in growth factors, such as Epidermal Growth Factor (EGF) and Connective Tissue Growth Factor (CTGF), which promote healing by leveraging the body’s natural healing mechanisms.
FDA Regulation and Research on PRP
Unlike HA, PRP is not FDA-regulated, and the devices used to prepare PRP are subject to FDA approval. Despite this, numerous studies have demonstrated PRP’s efficacy in treating tendon injuries and osteoarthritis, as well as in reducing pain. Ongoing research is exploring its potential benefits for hair regrowth, cardiac muscle repair, and dermatologic rejuvenation.
Comparing HA Injections and PRP
Effectiveness and Safety
Studies indicate that PRP can be just as effective, if not more so, than HA injections. While HA is FDA-approved only for knee use, PRP is derived from the patient’s own blood, reducing the risk of infection and autoimmune reactions, thanks to the presence of white blood cells that help fight infections.
Cost Considerations
HA injections for joints other than the knee are not FDA-approved or typically covered by insurance, often costing patients $1500 or more, excluding additional charges like doctor visits and the injection itself. In contrast, PRP treatments generally cost between $800 and $1200 out of pocket.
Conclusion: Which is the Better Choice?
PRP has demonstrated comparable or superior effectiveness to HA injections for arthritis pain management. It carries a lower risk of infection or autoimmune reactions and is generally more cost-effective. Given these advantages, PRP often emerges as the preferred choice for many patients and healthcare providers.
- Published in Corporate News / Blog
Platelet-Rich Plasma Stays Quietly Popular Despite Neglect

Fact: PRP Treatments Are Highly In-Demand
According to research, PRP treatments are one of the most in-demand treatments available in healthcare. This is impressive considering the following challenges PRP faces:
- Lack of Support from the Medical Industry: PRP is not backed by big pharma, meaning no extensive research or marketing is funded.
- Absence of Lobbying by Medical Associations: There are no medical associations working to increase awareness of PRP.
- No Insurance Reimbursements: Insurance companies do not reimburse PRP treatments, making it difficult to get patients to pay for a treatment that is relatively “unproven.”
- Rising Costs: In 2006, a PRP treatment cost $450. Today, it costs $800, with the cheapest being $650.
Despite these obstacles, the demand for PRP treatments remains robust.
The Future of PRP
We believe the best of PRP is yet to come. A breakthrough study could propel PRP into mainstream hospitals and clinics. The most significant growth in PRP is currently happening in Asia, rooted in fundamental healing theory.
The Healing Power of PRP
The growth of PRP can be attributed to its fundamental healing properties:
- Increased Platelets: More platelets mean more growth factors and cytokines, leading to enhanced healing.
- Natural Healing Mechanism: Our body’s natural healing mechanism operates with 150,000/ul-350,000/ul platelets in blood. Using PRP amplifies this number by 3X to 5X, translating to better healing.
Platelet-Rich Plasma Trends
PRP can be used to promote the healing of injured tendons, ligaments, muscles, and joints, and is applied to various musculoskeletal problems. Regular studies test its effectiveness. One landmark study involved double-blind randomized controlled trials to see the effect of PRP on patients with chronic low back pain caused by torn discs. The study found that 60% of the patients felt significant improvements, with some even being cured.
Platelet-Rich Plasma Variants
There are several types of PRP variants:
- Plasma Rich in Growth Factors (PRGF)
- Plasma Rich in Platelets and Growth Factors (PRPGF)
- Platelet-Rich Plasma (PRP)
- Platelet Poor Plasma (PPP)
- Plasma Rich in Platelets and Rich in Leukocytes (LR-PRP)
- Plasma Rich in Platelets and Poor in Leukocytes (LP-PRP)
- Platelet-Rich Fibrin Matrix (PRFM)
All of them involve plasmapheresis—the two-stage centrifugation process to separate platelets from blood. The industry has yet to standardize which variant to use, but we believe the confusion will clear up in 3-5 years.
Bio-Factors at Play in PRP
Regardless of the PRP variant used, the following bio-factors are at play:
- Growth Factors: TGF-B, PDGF, IGF-I,II, FGF, EGF, VEGF, ECGF
- Adhesive Proteins: Fibrinogen, Fibronectin, Vitronectin, Thrombospondin-1
- Clotting & Anti-Clotting Factors: Proteins, Antithrombin, Plasminogen, Proteases, Antiproteases
How Platelet-Rich Plasma Actually Works
PRP is commonly used for wound healing and pain management because platelets aid coagulation, act as a biological glue, and support stem or primary cell migration. They also help restore hyaluronic acid, accelerate collagen synthesis, and increase cartilage matrix.
Platelets delivered in a clot can immediately act as a scaffold to enable the healing process. 95% of bio-active proteins are released within 1 hour of injecting PRP, and platelets continue to release growth factors for 7-10 days. Thus, it’s recommended to re-inject PRP every 7 days.
Why Patients Choose PRP Despite the Cost
Patients are willing to pay out of pocket for PRP treatments despite insurance companies not covering it because:
- Effectiveness: The treatment works.
- Natural and Side-Effect-Free: There is nothing else as natural and side-effect-free as PRP.
PRP for Osteoarthritis
Consider osteoarthritis, which affects 27 million Americans. When doctors started doing PRP treatments for their osteoarthritis patients, a large majority experienced no further cartilage loss. This suggests PRP should be the default first-line treatment for osteoarthritis across the country.
PRP for Hair Loss and Cosmetic Applications
PRP also has significant potential in treating hair loss and cosmetic facial applications. Studies have shown reduced hair loss and increased hair density, with patients expressing high satisfaction levels.
The Growing PRP Market
The PRP market is expected to hit $126 million in 2016, a 180% increase over the 2009 figure of $45 million. For osteoarthritis alone, if all 27 million Americans received 1 PRP shot a year at a conservative $400 per treatment, it would be a $10 billion market.
PRP for Tennis Elbow
PRP is also known to work well for Tennis Elbow, which affects 1% to 3% of the overall population and up to 50% of tennis players.
Insurance Coverage for PRP
Getting PRP covered by insurance could significantly expand the market and help heal millions of patients more naturally and effectively. It could also save insurance companies money by reducing the need for more expensive interventions like surgery.
Challenges and the Future of PRP
The vast scope of PRP treatment calls for urgent structure and guidelines. There are some 20+ conditions where researchers have found it helpful. Proving its efficiency in all these areas is a daunting task, but we will get there with enough funding and standardized procedures.
Conclusion
Despite being classified as “unproven,” PRP has vast potential. With the right number of platelets, platelet activation, and cytokine release, PRP can consistently deliver positive outcomes. Although there is still uncertainty over the number of injections, timing, and delivery method, widespread adoption will eventually lead to a more structured approach.
Let’s hope the first glimpses of this structure will arrive soon. In 2015, the world saw approximately 1 million knee arthroplasties for osteoarthritis, costing $25 billion. How many of these patients could have benefited from PRP early on?
- Published in Corporate News / Blog
Platelet-Rich Plasma Injections: Protocol Guide

Almost all sports medicine doctors agree that there’s no harm in trying Platelet Rich Plasma Injections (PRP Injections) for their patients. After all, there are hundreds of thousands of cases with positive results. All it needs is research to prove its worth. Currently, many independent researches are ongoing, funded privately, like the one conducted by Dr. Kimberly G. Harmon M.D., director of the Primary Care Sports Medicine fellowship at the University of Washington. She recently received a gift to support her research from UW alumni who firmly believe in Platelet-Rich Plasma (PRP).
While the process of extracting PRP is fairly simple—there are many variants as long as platelets are above baseline levels with at least seven growth factors—many physicians are still unsure about what they can and can’t do when it comes to this marvelous procedure. Today, let’s shed light on the fine print.
Platelet-Rich Plasma Injections Protocol
Protocol/Technique
Usually, the procedure requires the physician/surgeon and an assistant or two to help with the preparation of the graft, maintenance of sterile technique, and saving the ultrasound images (if relevant).
Pre-Procedure Considerations

- There should always be a specific indication associated with a physical exam with confirmed imaging studies such as an ultrasound, CT scan, or MRI before treatment.
- Proper patient education and a discussion must be had with the patient, as well as a signed informed consent prior to the procedure.
- Contraindications must be reviewed prior to the procedure.
Graft Preparation
- The patient should be positioned in a comfortable seated or reclining position.
- Sterile single needles and syringes must be used with proper handling and disposal.
- Using an aseptic procedure, the proper amount of blood is then drawn from the vein for the PRP procedure.
- If the blood cannot be obtained from the site the first time, a new site must be used to prevent early activation.
- Using a sterile technique, transfer the tube of venous blood to the centrifuge. Platelet-Rich Plasma should be acquired using a separating device created for autologous blood. Preference is always given to a closed system that will prevent exposure of the blood and its cellular components to the open air, and permits minimal use of the tissue.
Image Guidance PRP Therapy
- Real-time imaging guidance using ultrasound, CT, or fluoroscopy should always be used when performing a PRP injection.
- If ultrasound is going to be used, the following considerations need to be decided in advance: For lengthy procedures, PRP injections near the spine, and intra-articular injections, sterile gel is recommended.
- Always use sterile probe covers. Cleansing the probe before and after the PRP procedures and observing sterile technique is sufficient.
- Guided images and indelible markings of the site of the probe position and the needle entry always need to be made before cleaning the skin where the probe and needle will be inserted.
- Always apply a bandage or a dressing after the procedure to protect the entry site from germs.
Post-Injection Care
- The patient should be monitored for any post-PRP procedure complications such as vaso-vagal reactions.
- Patients should be given post-procedure directions and precautions, and any questions should be answered before they leave. They should also have emergency contact information.
- Patients should be instructed about immobilization and any post-procedure activities that are allowed and/or not allowed.
- Post-PRP procedure pain prescriptions need to be given to the patient before discharge, and any questions about the medication(s) should be answered at this time. Patients also need to be instructed to avoid NSAIDs until they have healed, are pain-free, and have full function returned to the treated area.
- Per OSHA guidelines, contaminated areas must be disinfected before the next patient uses the room.
- The PRP procedure must be documented in detail, including a procedure note with the following information: date, pre- and post-procedure diagnosis, name of the procedure, physician/surgeon(s), any assistants, whether or not anesthesia was used and what type, short-term indication of the procedure, a description of the graft preparation, and a description of the procedure, including any/all guidance and instruments used.
Follow-up Care
- Patients are normally re-examined 2-6 weeks after the PRP procedure to follow up on pain, use, the injection site, and to discuss any concerns and future course of action.
- The patient response to the treatment should be recorded using authenticated outcome measures.
- Any complications, responses, and all other relevant information should be logged into the ICMS tracking system.
- Consideration for another PRP injection should be the center of the discussion, and the patient will be able to make a decision based on the outcome.
Safety Considerations
- Universal precautions must be used before, during, and after the procedure.
- Risk of infection: PRP is antimicrobial and provides effective protection against most bacterial infections except for Klebsiella, Pseudomonas, and Enterococcus.
- With the graft being made entirely out of autologous blood, it virtually eliminates the risk of disease transmission unless the graft becomes contaminated.
Risks to Patients from the Procedure
- Infection
- Bleeding
- Nerve damage
- Pain
- Lack of result
- Loss of limb and death (very rare)
Platelet-Rich Plasma: Indications
Musculoskeletal complaints require a complete history and exam to find a diagnosis. Often, diagnostic studies may be needed and reviewed to understand why prior treatments failed. PRP is usually considered an optional treatment for chronic and subacute conditions. Commonly, healing slows down or stops altogether at the 6-12 week period following an acute or traumatic injury. If the patient has not had any improvement for over the first six weeks, it’s probable the healing period has stopped.
Platelet-Rich Plasma: Contraindications
- Septicemia
- Platelet dysfunction syndrome
- Localized infection at the procedure site
- Hemodynamic instability
- Critical thrombocytopenia
- Patient not willing to take the risks involved with the procedure
Relative Contraindications:
- Regular use of NSAIDs within 48 hours of the PRP procedure
- HGB of < 10 g/dl
- Platelet count of < 105/ul
- Systemic use of corticosteroids within 2 weeks
- Recent illness or fever
- Cancer, particularly hematopoietic or bone
- HGB < 10 g/dl
- Platelet count < 105/ul
- Corticosteroid injection at the treatment site within 1 month
- Tobacco use
- Published in Corporate News / Blog
Platelet-Rich Plasma For Melasma — Will It Fade Forever?

For most women, a tiny pimple on the face is enough to ruin their day. Or week. Even the slightest imperfection that may have a 1% chance of getting noticed by others will freak them out. For these women, Melasma is their darkest nightmare. It’s a pretty common issue, a result of exposure to the sun, that causes brown patches on the face. Permanent patches, I should add.
If you’re suffering from Melasma, the road to “recovery” usually looks like this:
- You hope that it’ll fade away.
- Your friend suggests you try apple cider vinegar and lemon juice treatment.
- Slightly disappointed.
- You visit a dermatologist who’ll prescribe a bleaching cream (hydroquinone or similar).
- Full-on disappointment.
- You Google the hell out of the topic.
- Overwhelm.
- Concealers and makeup become your best friend.
At this point, no one can convince you there is a treatment for getting rid of melasma. Trying more and more treatments only runs the risk of making the condition worse. So what would you do?
What About Platelet-Rich Plasma For Melasma?
According to recent Turkish and Malaysian studies, Platelet-Rich Plasma is showing great promise for melasma. The one good thing about PRP for Melasma is the fact that PRP won’t make the condition worse unlike IPL, Fraxel, or other treatments. So that’s one of the treatments you can confidently try without worry. It’s like getting a natural facial treatment that has a whole lot of potential benefits even if it didn’t help cure melasma.
PRP injections work by supplying growth factors to reduce the pigmentation. And being an independent treatment with no downtime, it can be done in conjunction with conventional treatments for melasma to add and enhance the effects. There are more than 30 bioactive substances in Platelet-Rich Plasma that have separate roles like increasing skin volume and adding new blood vessels to name a few.
Platelet-Rich Plasma with Microneedling
This is the most common combination for Platelet-Rich Plasma therapy. Here’s a video of Dr. Michael Somenek performing a PRP injection on a patient of his immediately after microneedling. The combination is known to have produced results for a lot of varieties of skin pigmentation issues that it’d not be wise for anyone to ignore it for melasma, especially when creams and peels didn’t help. More important is PRP’s ability to stimulate collagen production in the area so it tightens the pores and makes your skin glowing.
Why Platelet-Rich Plasma?
PRP is primarily a healing vehicle. It needs to be injected into the membrane below the skin. The way it works is by supplying the underlying skin membrane with collagen and tenascin stimulated by the transforming growth factors in PRP. These growth factors also promote the formation of new blood vessels that in some cases results in the disappearance of spider veins.
The released growth factors (mainly platelet-derived growth factor (PDGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and transforming growth factor-beta (TGF-ß)) can stimulate the proliferation of fibroblasts and epidermal cells, and collagen synthesis. In addition, the transforming growth factor-beta (TGF-ß) has been proven to inhibit melanogenesis — or reverse skin pigmentation — the exact opposite effect of exposure to UV-B radiation.
Typically, patients see excellent results with 2-3 PRP injections in the first 3 months. And clinical studies have shown that it will maintain after 6 months. However, Melasma is known to recur even after successful treatments. So you must take precautions against it by using sunscreen with broad-spectrum protection and an SPF of 30 or higher. And avoid skincare products that are harsh as they can exacerbate melasma.
- Published in Corporate News / Blog
Platelet-Rich Plasma For Melasma — Will It Fade Forever?

For most women, a tiny pimple on the face is enough to ruin their day. Or week. Even the slightest imperfection that may have a 1% chance of getting noticed by others will freak them out. For these women, Melasma is their darkest nightmare. It’s a pretty common issue, a result of exposure to the sun, that causes brown patches on the face. Permanent patches, I should add.
If you’re suffering from Melasma, the road to “recovery” usually looks like this:
- You hope that it’ll fade away.
- Your friend suggests you try apple cider vinegar and lemon juice treatment.
- Slightly disappointed.
- You visit a dermatologist who’ll prescribe a bleaching cream (hydroquinone or similar).
- Full-on disappointment.
- You Google the hell out of the topic.
- Overwhelm.
- Concealers and makeup become your best friend.
At this point, no one can convince you there is a treatment for getting rid of melasma. Trying more and more treatments only runs the risk of making the condition worse. So what would you do?
What About Platelet-Rich Plasma For Melasma?
According to recent Turkish and Malaysian studies, Platelet-Rich Plasma is showing great promise for melasma. The one good thing about PRP for Melasma is the fact that PRP won’t make the condition worse unlike IPL, Fraxel, or other treatments. So that’s one of the treatments you can confidently try without worry. It’s like getting a natural facial treatment that has a whole lot of potential benefits even if it didn’t help cure melasma.
PRP injections work by supplying growth factors to reduce the pigmentation. And being an independent treatment with no downtime, it can be done in conjunction with conventional treatments for melasma to add and enhance the effects. There are more than 30 bioactive substances in Platelet-Rich Plasma that have separate roles like increasing skin volume and adding new blood vessels to name a few.
Platelet-Rich Plasma with Microneedling
This is the most common combination for Platelet-Rich Plasma therapy. Here’s a video of Dr. Michael Somenek performing a PRP injection on a patient of his immediately after microneedling. The combination is known to have produced results for a lot of varieties of skin pigmentation issues that it’d not be wise for anyone to ignore it for melasma, especially when creams and peels didn’t help. More important is PRP’s ability to stimulate collagen production in the area so it tightens the pores and makes your skin glowing.
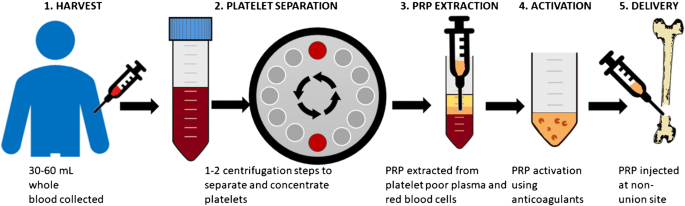
Why Platelet-Rich Plasma?
PRP is primarily a healing vehicle. It needs to be injected into the membrane below the skin. The way it works is by supplying the underlying skin membrane with collagen and tenascin stimulated by the transforming growth factors in PRP. These growth factors also promote the formation of new blood vessels that in some cases results in the disappearance of spider veins.
The released growth factors (mainly platelet-derived growth factor (PDGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and transforming growth factor-beta (TGF-ß)) can stimulate the proliferation of fibroblasts and epidermal cells, and collagen synthesis. In addition, the transforming growth factor-beta (TGF-ß) has been proven to inhibit melanogenesis — or reverse skin pigmentation — the exact opposite effect of exposure to UV-B radiation.
Typically, patients see excellent results with 2-3 PRP injections in the first 3 months. And clinical studies have shown that it will maintain after 6 months. However, Melasma is known to recur even after successful treatments. So you must take precautions against it by using sunscreen with broad-spectrum protection and an SPF of 30 or higher. And avoid skincare products that are harsh as they can exacerbate melasma.
- Published in Corporate News / Blog
tem Cell Platelet-Rich Plasma: The Best Regenerative Therapy?

To understand why stem cell platelet-rich plasma or co-transplantation of adipose-derived mesenchymal stem cells (ADSC) and PRP is such a remarkable idea in regenerative medicine, let’s spend a little time looking at the mechanics of PRP.
Platelet-Rich Plasma’s Role as Repairmen
The one thing that makes Platelet-Rich Plasma a hero in several fields (if not all) of medicine is the fact that the diverse growth factors in it are able to stimulate stem cell proliferation and cell differentiation (the factors that determine effective tissue regeneration and healing) on any part of the body. These growth factors are abundant in the blood and act as the natural repairmen of tissues. In the perfect scenario, there’s plenty of blood flow to every part of the body, and these “repairmen” are always on-call to address any healing needs that may arise. However, if the injured area has a poor blood supply — especially areas that constantly move like tendons, ligaments, and joints — demand for these repairmen can outgrow supply. Meaning, healing (or regeneration of tissues) is put on hold till further repairmen are available.
The train of Platelet-Rich Plasma then arrives with enough of these repairmen to warrant the resumption of healing. There’s another part of this picture we haven’t talked about so far: stem cells.
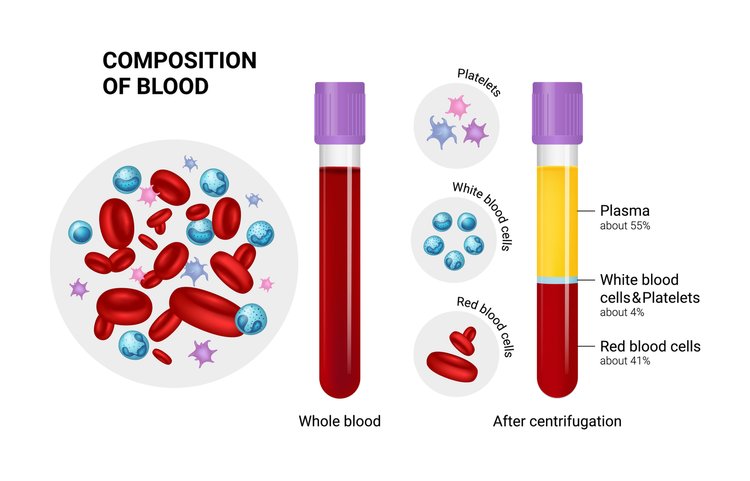
Stem Cells as the Raw Materials for PRP
As far as Platelet-Rich Plasma and its growth factors are concerned, they are mere repairmen. They can’t do the work by themselves. They need the basic raw materials to work with. And that raw material here is stem cells. Stem cells are the ones actually being regenerated to form new tissues for healing.
Supplying Both PRP and Stem Cells for Regeneration
Stem cells are the only raw materials that PRP works with for regeneration. These are like the fundamental building blocks of all other cells. These cells can be guided into becoming specialized cells under the right conditions. In addition, they can also divide themselves to form new stem cells or new specialized cells. So for Platelet-Rich Plasma to work well, it needs to be applied to an area with lots of stem cells like the heart, liver, blood vessels, etc. Incidentally, Platelet-Rich Plasma’s healing properties were first discovered by cardiac surgeons who played with concentrated blood for faster healing of the heart after surgery, and it showed tremendous promise because stem cells are abundant in heart tissues. But what if healing is needed in an area where there are not many stem cells?
With the new developments in stem cell technology, that can be solved too. Because now we can supply the stem cells to areas where there are less like the joints, ligaments, and tendons. For this, scientists usually use “mesenchymal stem cell” or MSCs. These are cells isolated from stroma and can differentiate to form adipocytes, cartilage, bone, tendons, muscle, and skin. The easiest way is to harvest it from adipose tissue or fat that we call Adipose-derived mesenchymal stem cells or ADSC.
The Future of Stem Cell Platelet-Rich Plasma
In regions with hypoxia (poor blood supply) like joints, meniscus tissue, rotator cuff, spinal discs, etc., the supply of platelets (and therefore growth factors) as well as the stem cells are limited. So what if we supplied both the stem cells and Platelet-Rich Plasma for triggering the regeneration process? That’s the question these Japanese scientists answered in their research. Here’s another group of scientists who took on the same challenge.
They used Adipose-derived mesenchymal stem cells (ADSC), which are known for their ease of isolation and extensive differentiation potential. These researchers noted that these stem cells often can’t survive in areas of local hypoxia, oxidative stress, and inflammation – thereby making them ineffective. However, when Platelet-Rich Plasma (or thrombin-activated PRP) is added to ADSC, it kept them alive for prolonged periods and the growth factors in the Platelet-Rich Plasma triggered cell differentiation and proliferation more easily.
Why This Exact Combination Is the Future
Done this way, both Adipose-derived mesenchymal stem cells (ADSC) and Platelet-Rich Plasma are raw materials for healing that’s already available in plenty in almost everyone (there are exceptions of course). That means, for complete healing to take place, this combination treatment, still in its very primitive stage of development, may have the potential to replace expensive synthetic drugs that carry complex unexplained side effects. The procedure takes our body’s natural healing agents — stem cells from body fat and PRP from blood — and then injects them inside the knee or other joints (or other areas where they are insufficient) for regeneration. Isn’t that like the most wonderful thing ever?
Whether it’s a cartilage cell, a bone cell, or a collagen cell for ligaments and tendons that need to be healed, all you need is a same-day procedure by a local, but specialized doctor, using the natural ingredients of the body. I believe this special combo is a huge win for Platelet-Rich Plasma.
The Challenges for Growing Adoption of This Treatment
We know Platelet-Rich Plasma has safe, yet high-speed recovery potential with its multiple growth factors. And it is effective in regenerative healing of cartilage injuries – the toughest injuries to heal – as well as Osteoarthritis. However, the challenges are Platelet Quality. We need to somehow ensure the Platelet-Rich Plasma quality is uniform. Currently, it varies from two to several fold above baseline concentration based on the donor’s physical condition.
Next, we need to identify the exact PRP growth factors that promote ADSC proliferation. Scientists believe growth factors such as basic fibroblast growth factor (bFGF), epidermal growth factor, and platelet-derived growth factor stimulate stem cell proliferation while some growth factors under certain conditions are known to inhibit the process. The percentage of PRP matters too. Scientists have tested 5 percent, 10 percent, 15 percent, and 20 percent Platelet-Rich Plasma in ADSC.
The Only Treatment in Modern Medicine for Cartilage Regeneration
The bottom line is that Stem Cell Platelet-Rich Plasma or ADSC + PRP procedure is the only treatment in modern medicine that has shown cartilage regeneration. So it’s too important to ignore. And it could be one of the greatest advances that science has brought to the millions of people suffering from serious pain in their joints, knee, and spine as well as people suffering from all kinds of tendon diseases and injuries.
To understand why stem cell platelet-rich plasma or co-transplantation of Adipose-derived mesenchymal stem cells and PRP, is such a remarkable idea in regenerative medicine, let’s spend a little time looking at the mechanics of PRP.
- Published in Corporate News / Blog
Engineering Stem and Stromal Cell Therapies for Musculoskeletal Tissue Repair

Musculoskeletal tissue injuries and degeneration are common and debilitating for a high number of patients (Brooks, 2006). Unfortunately, endogenous musculoskeletal tissue regeneration is limited in many cases and may be affected by inflammation and the degree of damage. For example, most fractures of long bones heal spontaneously, whereas large segmental defects fail to heal. Additionally, although articular cartilage has almost no intrinsic reparative potential, tendons and ligaments may heal, but often with inferior properties. The high prevalence of these injuries has led to significant investment in the development of new therapies to enhance healing and augment current surgical interventions. Often the goal is to mimic and recapitulate the natural healing cascade and developmental process by transplantation of tissue-specific stromal and progenitor cells or by endogenous manipulation to enhance the native repair capacity of cells.
Advances in Stem and Stromal Cell Therapies
There has been a continuing increase in the number and type of stem and stromal cells being pursued in human clinical trials for treatment of musculoskeletal injuries (Steinert et al., 2012). Most approaches in this area are based on ex vivo-expanded mesenchymal stromal cells (MSCs) derived from bone marrow (BM). Originally identified and characterized by their multilineage differentiation potential in vitro, multipotent capabilities of MSCs in vivo have not been clearly demonstrated to date, particularly because of the lack of methods to identify and define differentiated populations (Nombela-Arrieta et al., 2011). Central to recent progress in the field has been the understanding that stem and progenitor functions of MSCs may not be the key attribute that mediates tissue repair. In addition, there is outstanding controversy over the terminology of exogenously supplied MSCs as stromal cells, and various terms, including medicinal signaling cells, have been proposed to more accurately reflect their therapeutic function in vivo (Caplan, 2017). Nevertheless, the therapeutic benefit of these cells has been largely explored. Significant advances have been made in developing strategies that deliver, protect, and recruit stem cells, and the bioengineering field is evolving to improve current surgical techniques.
Current Treatments and Clinical Investigations
This review first describes current treatments and reports the recent progress in clinical investigations of stem and stromal cell-based therapies for musculoskeletal repair with a particular focus on bone and fibrocartilaginous tissues. The current understanding of appropriate cell sources and delivery strategies is then illustrated toward endogenous repair of musculoskeletal tissues. Last, emerging therapeutic concepts are highlighted in the context of biomaterials as a particularly attractive tool to control stem and stromal cell behavior both ex vivo and in vivo, to recruit endogenous stem cells, and to control the local healing environment. Such approaches have great potential for future therapies in musculoskeletal repair.
Bone Repair
The intrinsic repair of bone defects mirrors many events of embryonic development and makes fracture healing one of the rare postnatal processes that are regenerative and can ultimately restore damaged tissue to its pre-injury structure, composition, and biomechanical function (Figure 1). In spite of the unique capacity of bone to heal, a number of clinical indications remain where therapeutic intervention is required. In the case of complex trauma with multiple fractures, infections, and tumor-associated and endocrine diseases (e.g., diabetes, osteoporosis), the body’s natural healing response is impaired, and non-union can occur in up to 15% of cases (Grayson et al., 2015). Another debilitating disorder is non-traumatic avascular osteonecrosis, which can lead to collapse of the femoral head and accounts for 10,000–20,000 total hip replacement surgeries in the United States per year (Figure 1; Moya-Angeler et al., 2015). Autologous bone grafting represents the gold standard for management of bone defects and non-unions, and union rates of more than 90% have been reported using iliac crest bone. However, considerable donor site morbidity and limited volumes must be taken into consideration. Additionally, allogeneic or synthetic bone substitutes, such as ceramics, corals, or polymer-based materials, have not reached the biological and mechanical properties equivalent to autologous bone (Table 1).
Skeletal Muscle
In addition to direct traumatic injury, complex damage of bone tissue (e.g., open fractures, tumor ablations) often results in concomitant soft tissue injury, including adjacent muscles. Although skeletal muscle has the inherent ability to regenerate after injuries, the regenerative capacity fails when a large volume of muscle is lost (i.e., volumetric loss). Such severe injuries may lead to fibrosis, atrophy, and ischemia when left untreated, accounting for significant socioeconomic costs ($18.5 billion in healthcare costs are associated with sarcopenia alone) (Janssen et al., 2004). Therapeutic treatment options are limited to physical therapy, scar tissue debridement, and transfer of healthy, innervated, and vascularized autologous muscle tissue. However, the outcomes of surgical reconstructions often remain aesthetically and functionally deficient (Grogan et al., 2011; Table 1).
Articular Cartilage and Meniscus
In contrast to bone and skeletal muscle tissue, the poor intrinsic healing capacity of articular cartilage and meniscus tissue presents a major challenge in clinics. Lesions from injuries or degeneration often result in gradual tissue erosion, leading to impaired function of the affected joint and degenerative osteoarthritis (OA) (Figure 1). Patients with post-traumatic OA account for more than 10% of the 27 million adults in the United States that have a clinical diagnosis of OA (Johnson and Hunter, 2014). Commonly, the first-line treatment of articular injuries includes arthroscopic lavage, partial meniscectomy, and BM stimulation techniques to penetrate subchondral bone (Table 1). Microfracture has been considered the gold standard for stimulating endogenous repair; however, it often results in the formation of inferior fibrocartilaginous repair tissue. This cartilaginous tissue is vulnerable due to altered biomechanics of the subchondral bone, which raises concerns about the long-term efficacy of microfracture (Solheim et al., 2016). Therefore, secondary and more complex procedures strive to restore the hyaline cartilage, such as osteochondral autografting from the less weight-bearing periphery (mosaicplasty) and autologous chondrocyte implantation (ACI). ACI represents one of the first clinical applications of tissue engineering where a biopsy from a low-weight-bearing region is performed, and ex vivo-expanded chondrocytes are implanted in a second operation. The de-differentiation of monolayer expanded chondrocytes and potential of recovery when implanted has been a matter of debate, and matrix-based ACI techniques have been developed that use absorbable scaffolds (e.g., porcine collagen) to support the implanted cells (Makris et al., 2015). An important limitation of these techniques is the long recovery time (6–12 months) to ensure neotissue formation. The choice of articular injury treatment depends on several factors, including localization and size of the lesion, the level of activity, and the degree of associated damage of menisci and ligaments.
Meniscus Repair
Tears of the fibrocartilaginous menisci require surgical intervention for nearly 1 million patients in the United States annually (Vrancken et al., 2013). For lesions located in the peripheral vascularized region of the meniscus, repair strategies such as sutures and anchors allow preservation of the meniscal tissue. However, meniscal lesions often appear in the avascular central regions, which makes them less suitable for healing and usually requires partial or (sub)total meniscectomy (Figure 1; Table 1). In some cases, further treatment with a meniscal substitute, such as an allograft or a synthetic implant, is indicated to limit OA (Vrancken et al., 2013).
Other Fibrous Musculoskeletal Tissues
Another large proportion of musculoskeletal injuries in the clinic is represented by other damaged fibrous structures, including tendons, ligaments, and the annulus fibrosus (AF). Often, degenerative pathology precedes acute trauma, and, like articular cartilage, these tissues have a limited healing capacity. One of the most common tendon injuries presented clinically is tearing of one or more of the interdigitating tendons of the rotator cuff (Figure 1). Failure of initial physical therapy or acute trauma in young patients motivates surgical repair using open or arthroscopic approaches for subacromial decompression, tendon debridement, and suture or anchor supplementation (Table 1). Still, repair is limited, particularly within the complex anatomic arrangement forming the shoulder cuff. The formation of fibrovascular scar tissue frequently leads to significant morbidity, re-ruptures, and difficulties in treatment choice.
The intervertebral discs (IVDs) are composed of the nucleus pulposus (NP), a hydrophilic proteoglycan-rich gelatinous core, surrounded by a dense fibrocartilage ring—the AF (Figure 1). The gradual progression of IVD degeneration and the extrusion of the NP through defects in the AF is a major cause for lower back pain, a leading cause of global disability (Sakai and Andersson, 2015). Available treatments are mostly symptomatic, and surgical treatments often resect the structural obstruction resulting from herniation or fuse motion segments (Table 1). However, the complex structural features of IVDs surrounded by neural elements and inflammation frequently cause a homeostatic imbalance favoring a catabolic response governed by the loss of the IVD structure, which is often followed by facet joint arthritis and vertebra deformation, canal stenosis, and even deformations. Most importantly, disc replacement with synthetic implants or fusion of the motion segment does not cure the underlying pathology of IVD degeneration (Sakai and Andersson, 2015).
- Published in Corporate News / Blog
